

zol and fai: large-scale targeted detection and evolutionary investigation of gene clusters
- PMID: 39907107
- PMCID: PMC11795205
- DOI: 10.1093/nar/gkaf045
zol and fai: large-scale targeted detection and evolutionary investigation of gene clusters
Abstract
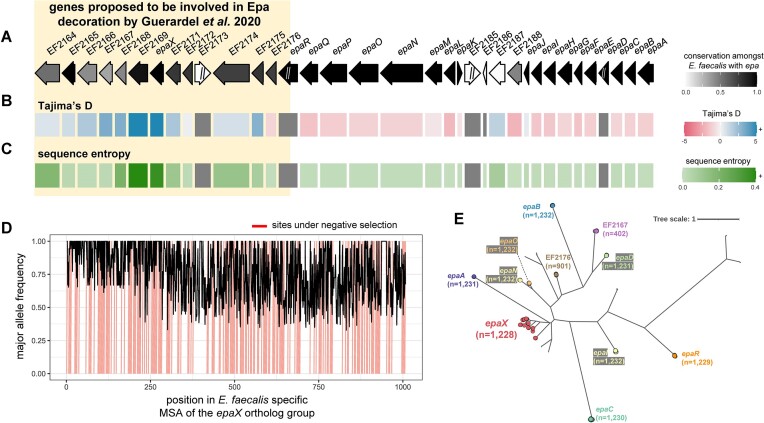
Many universally and conditionally important genes are genomically aggregated within clusters. Here, we introduce fai and zol, which together enable large-scale comparative analysis of different types of gene clusters and mobile-genetic elements, such as biosynthetic gene clusters (BGCs) or viruses. Fundamentally, they overcome a current bottleneck to reliably perform comprehensive orthology inference at large scale across broad taxonomic contexts and thousands of genomes. First, fai allows the identification of orthologous instances of a query gene cluster of interest amongst a database of target genomes. Subsequently, zol enables reliable, context-specific inference of ortholog groups for individual protein-encoding genes across gene cluster instances. In addition, zol performs functional annotation and computes a variety of evolutionary statistics for each inferred ortholog group. Importantly, in comparison to tools for visual exploration of homologous relationships between gene clusters, zol can scale to handle thousands of gene cluster instances and produce detailed reports that are easy to digest. To showcase fai and zol, we apply them for: (i) longitudinal tracking of a virus in metagenomes, (ii) performing population genetic investigations of BGCs for a fungal species, and (iii) uncovering evolutionary trends for a virulence-associated gene cluster across thousands of genomes from a diverse bacterial genus.
© The Author(s) 2025. Published by Oxford University Press on behalf of Nucleic Acids Research.
Conflict of interest statement
None declared.
Figures

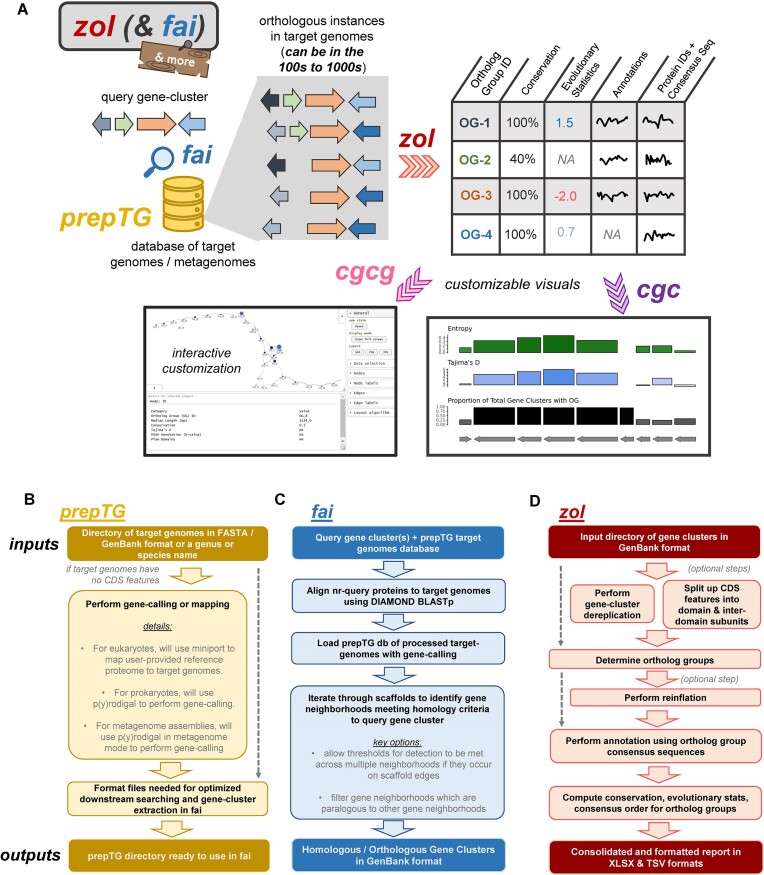
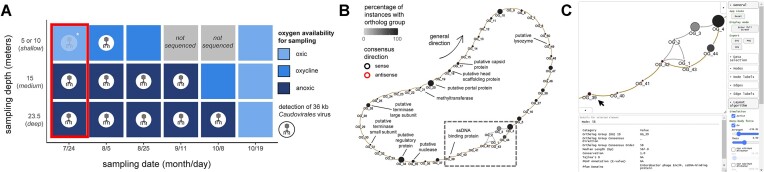
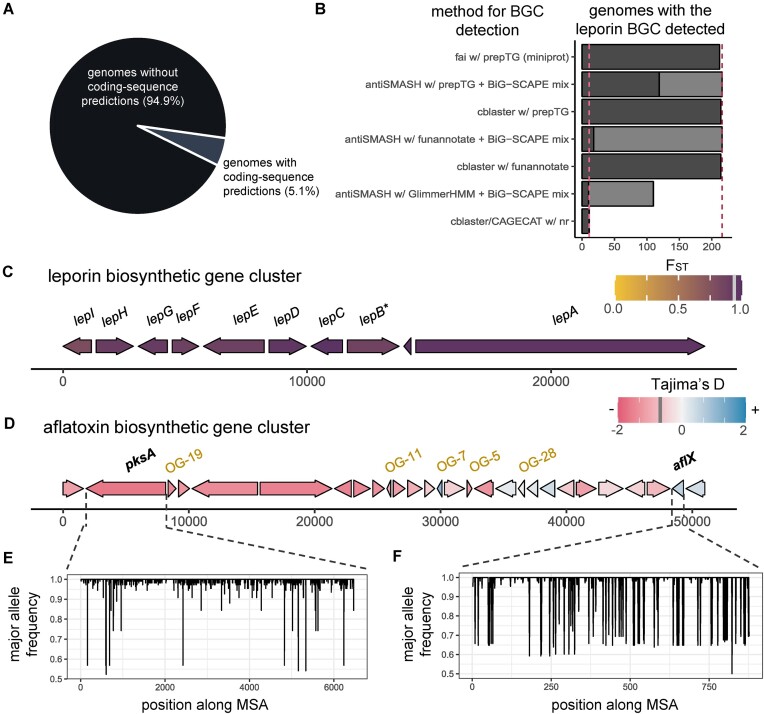
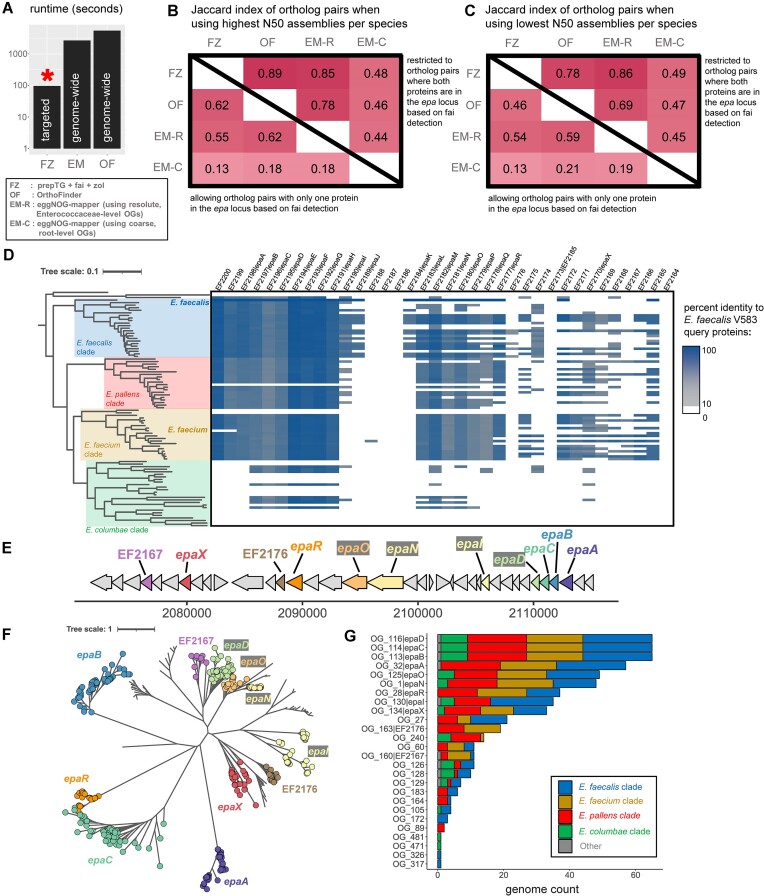
Update of
-
zol & fai: large-scale targeted detection and evolutionary investigation of gene clusters.bioRxiv [Preprint]. 2024 Sep 12:2023.06.07.544063. doi: 10.1101/2023.06.07.544063. bioRxiv. 2024. Update in: Nucleic Acids Res. 2025 Jan 24;53(3):gkaf045. doi: 10.1093/nar/gkaf045. PMID: 37333121 Free PMC article. Updated. Preprint.
Similar articles
-
zol & fai: large-scale targeted detection and evolutionary investigation of gene clusters.bioRxiv [Preprint]. 2024 Sep 12:2023.06.07.544063. doi: 10.1101/2023.06.07.544063. bioRxiv. 2024. Update in: Nucleic Acids Res. 2025 Jan 24;53(3):gkaf045. doi: 10.1093/nar/gkaf045. PMID: 37333121 Free PMC article. Updated. Preprint.
-
FUNYBASE: a FUNgal phYlogenomic dataBASE.BMC Bioinformatics. 2008 Oct 27;9:456. doi: 10.1186/1471-2105-9-456. BMC Bioinformatics. 2008. PMID: 18954438 Free PMC article.
-
MIBiG 2.0: a repository for biosynthetic gene clusters of known function.Nucleic Acids Res. 2020 Jan 8;48(D1):D454-D458. doi: 10.1093/nar/gkz882. Nucleic Acids Res. 2020. PMID: 31612915 Free PMC article.
-
Microbial genome analysis: the COG approach.Brief Bioinform. 2019 Jul 19;20(4):1063-1070. doi: 10.1093/bib/bbx117. Brief Bioinform. 2019. PMID: 28968633 Free PMC article. Review.
-
The quest for orthologs: finding the corresponding gene across genomes.Trends Genet. 2008 Nov;24(11):539-51. doi: 10.1016/j.tig.2008.08.009. Epub 2008 Sep 24. Trends Genet. 2008. PMID: 18819722 Review.
Cited by
-
Large-scale investigation for antimicrobial activity reveals novel defensive species across the healthy skin microbiome.bioRxiv [Preprint]. 2024 Nov 4:2024.11.04.621544. doi: 10.1101/2024.11.04.621544. bioRxiv. 2024. PMID: 39574598 Free PMC article. Preprint.
-
skDER & CiDDER: two scalable approaches for microbial genome dereplication.bioRxiv [Preprint]. 2025 Mar 6:2023.09.27.559801. doi: 10.1101/2023.09.27.559801. bioRxiv. 2025. Update in: Microb Genom. 2025 Jul;11(7). doi: 10.1099/mgen.0.001438. PMID: 38045253 Free PMC article. Updated. Preprint.
-
Context matters: assessing the impacts of genomic background and ecology on microbial biosynthetic gene cluster evolution.mSystems. 2025 Mar 18;10(3):e0153824. doi: 10.1128/msystems.01538-24. Epub 2025 Feb 24. mSystems. 2025. PMID: 39992097 Free PMC article. Review.
-
Targeted genome mining with GATOR-GC maps the evolutionary landscape of biosynthetic diversity.bioRxiv [Preprint]. 2025 Feb 28:2025.02.24.639861. doi: 10.1101/2025.02.24.639861. bioRxiv. 2025. Update in: Nucleic Acids Res. 2025 Jul 8;53(13):gkaf606. doi: 10.1093/nar/gkaf606. PMID: 40060561 Free PMC article. Updated. Preprint.
-
Targeted genome mining with GATOR-GC maps the evolutionary landscape of biosynthetic diversity.Nucleic Acids Res. 2025 Jul 8;53(13):gkaf606. doi: 10.1093/nar/gkaf606. Nucleic Acids Res. 2025. PMID: 40626555 Free PMC article.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
