

Combinatorial In Vivo Genome Editing Identifies Widespread Epistasis and an Accessible Fitness Landscape During Lung Tumorigenesis
- PMID: 39907430
- PMCID: PMC11824425
- DOI: 10.1093/molbev/msaf023
Combinatorial In Vivo Genome Editing Identifies Widespread Epistasis and an Accessible Fitness Landscape During Lung Tumorigenesis
Abstract
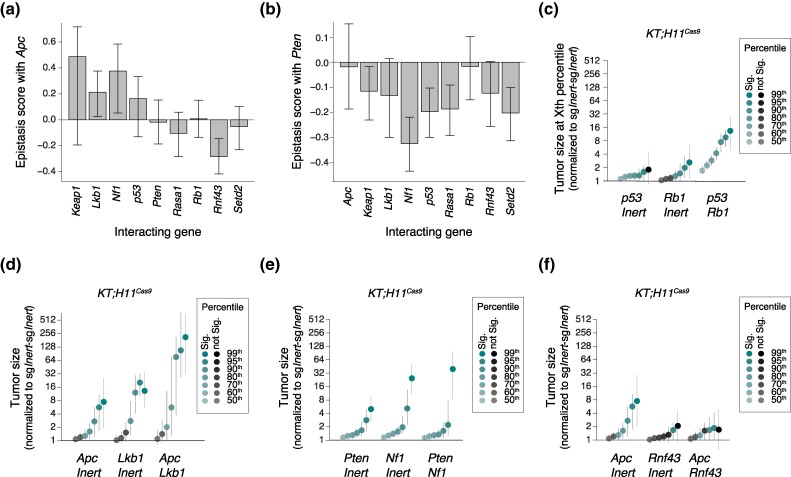
Lung adenocarcinoma, the most common subtype of lung cancer, is genomically complex, with tumors containing tens to hundreds of non-synonymous mutations. However, little is understood about how genes interact with each other to enable the evolution of cancer in vivo, largely due to a lack of methods for investigating genetic interactions in a high-throughput and quantitative manner. Here, we employed a novel platform to generate tumors with inactivation of pairs of ten diverse tumor suppressor genes within an autochthonous mouse model of oncogenic KRAS-driven lung cancer. By quantifying the fitness of tumors with every single and double mutant genotype, we show that most tumor suppressor genetic interactions exhibited negative epistasis, with diminishing returns on tumor fitness. In contrast, Apc inactivation showed positive epistasis with the inactivation of several other genes, including synergistic effects on tumor fitness in combination with Lkb1 or Nf1 inactivation. Sign epistasis was extremely rare, suggesting a surprisingly accessible fitness landscape during lung tumorigenesis. These findings expand our understanding of the interactions that drive tumorigenesis in vivo.
Keywords: cancer evolution; cancer models; epistasis.
© The Author(s) 2025. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures



Update of
-
Combinatorial in vivo genome editing identifies widespread epistasis during lung tumorigenesis.bioRxiv [Preprint]. 2024 Mar 8:2024.03.07.583981. doi: 10.1101/2024.03.07.583981. bioRxiv. 2024. Update in: Mol Biol Evol. 2025 Feb 03;42(2):msaf023. doi: 10.1093/molbev/msaf023. PMID: 38496564 Free PMC article. Updated. Preprint.
References
-
- Alonso-del Valle A, León-Sampedro R, Rodríguez-Beltrán J, DelaFuente J, Hernández-García M, Ruiz-Garbajosa P, Cantón R, Peña-Miller R, San Millán A. Variability of plasmid fitness effects contributes to plasmid persistence in bacterial communities. Nat Commun. 2021:12(1):2653. 10.1038/s41467-021-22849-y. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
