

A neoantigen vaccine generates antitumour immunity in renal cell carcinoma
- PMID: 39910301
- PMCID: PMC11903305
- DOI: 10.1038/s41586-024-08507-5
A neoantigen vaccine generates antitumour immunity in renal cell carcinoma
Abstract
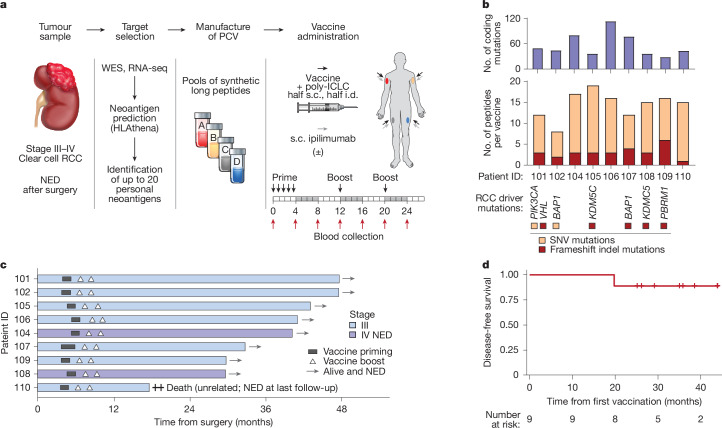
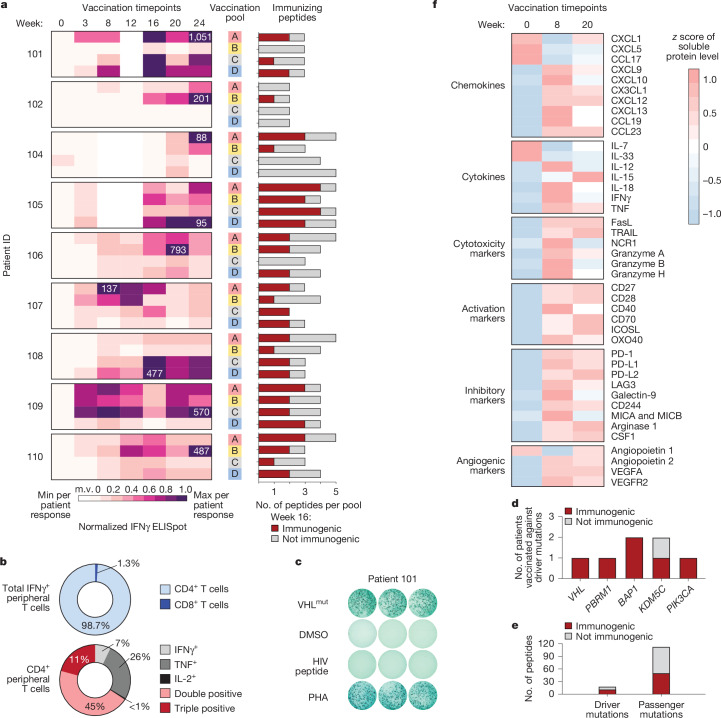
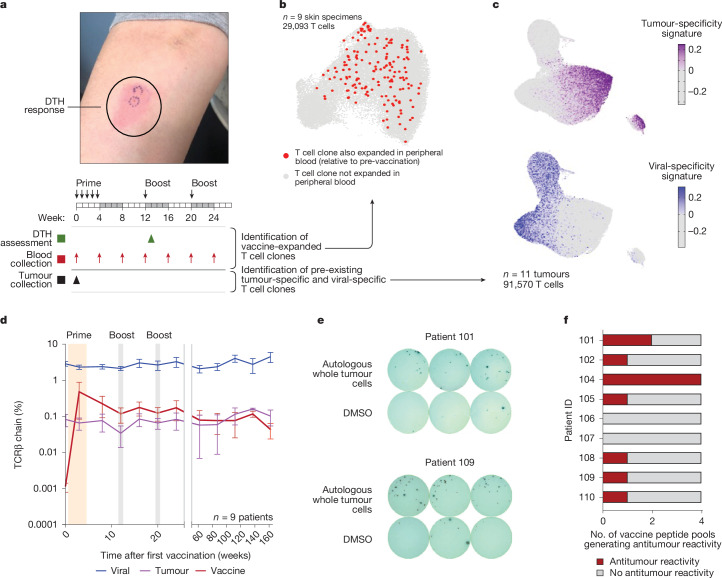
Personalized cancer vaccines (PCVs) can generate circulating immune responses against predicted neoantigens1-6. However, whether such responses can target cancer driver mutations, lead to immune recognition of a patient's tumour and result in clinical activity are largely unknown. These questions are of particular interest for patients who have tumours with a low mutational burden. Here we conducted a phase I trial (ClinicalTrials.gov identifier NCT02950766) to test a neoantigen-targeting PCV in patients with high-risk, fully resected clear cell renal cell carcinoma (RCC; stage III or IV) with or without ipilimumab administered adjacent to the vaccine. At a median follow-up of 40.2 months after surgery, none of the 9 participants enrolled in the study had a recurrence of RCC. No dose-limiting toxicities were observed. All patients generated T cell immune responses against the PCV antigens, including to RCC driver mutations in VHL, PBRM1, BAP1, KDM5C and PIK3CA. Following vaccination, there was a durable expansion of peripheral T cell clones. Moreover, T cell reactivity against autologous tumours was detected in seven out of nine patients. Our results demonstrate that neoantigen-targeting PCVs in high-risk RCC are highly immunogenic, capable of targeting key driver mutations and can induce antitumour immunity. These observations, in conjunction with the absence of recurrence in all nine vaccinated patients, highlights the promise of PCVs as effective adjuvant therapy in RCC.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: Bristol-Myers Squibb provided ipilimumab, but no funding support for the clinical trial. D.A.B reports share options in Elephas; advisory board, consulting or personal fees from Cancer Expert Now, Adnovate Strategies, MDedge, CancerNetwork, Catenion, OncLive, Cello Health BioConsulting, PWW Consulting, Haymarket Medical Network, Aptitude Health, ASCO Post and Harborside, Targeted Oncology, Merck, Pfizer, MedScape, Accolade 2nd MD, DLA Piper, AbbVie, Compugen, Link Cell Therapies, Scholar Rock, NeoMorph, Nimbus, Exelixis, AVEO, Eisai and Elephas; and research support from Exelixis and AstraZeneca, outside the submitted work. B.A.M. discloses personal consulting fees from Arcus, Aveo, Bristol-Myers Squibb, Daiichi Sanko, Eisai, Exelixis, Genmab, Gilead, Hexagen, Pfizer and SeaGen, and institutional disclosures for Aveo, Bristol-Myers Squibb, Exelixis, Gilead, Pfizer and SeaGen. N.R.L. is a consultant and has received honoraria from Bayer, Seattle Genetics, Sanofi, Silverback, Fortress Biotech, Synox Therapeutics, Janssen and Astellas pharma, outside the submitted work. K.S. is now an employee and shareholder of Intellia Therapeutics, which has no contribution or affiliation to the work described in this article. S.K. is now an employee of Genentech. S. Sarkizova is now an employee of Moderna and holds stock (as of January 2024). J.B.I. reports receiving consulting fees from AstraZeneca outside the submitted work. S. Signoretti reports receiving commercial research grants from Bristol-Myers Squibb, AstraZeneca, Exelixis, Merck, NiKang Therapeutics and Arsenal Biosciences; is a consultant/advisory board member for Merck, AstraZeneca, Bristol Myers Squibb, CRISPR Therapeutics AG, AACR and NCI; receives royalties from Biogenex; and mentored several non-United States citizens on research projects with potential funding (in part) from non-United States sources/Foreign Components. J.C.A. has served as a consultant for Remix Therapeutics, Ayala Pharmaceuticals, Cellestia, and on the scientific advisory board of NeuAPC, outside the submitted work. S.A.C. is a member of the scientific advisory boards of PTM BioLabs, Seer, StandUp2Cancer and PrognomIQ. I.L. serves as a consultant for PACT Pharma and has stock, is on the board and serves as a consultant for ennov1, and is on the board and holds equity in Nord Bio. G.G. receives research funds from IBM, Pharmacyclics/Abbvie, Bayer, Genentech, Calico, Ultima Genomics, Inocras and Google and is also an inventor on patent applications filed by the Broad Institute related to MSMuTect, MSMutSig, POLYSOLVER, SignatureAnalyzer-GPU, MSEye and MinimuMM-seq. He is a founder, consultant and holds privately held equity in Scorpion Therapeutics; he is also a founder of, and holds privately held equity in, PreDICTA Biosciences. He was also a consultant to Merck. N.H. holds equity in and advises Danger Bio/Related Sciences and Repertoire Immune Medicines, owns equity in BioNtech and receives research funding from Bristol Myers Squibb and Calico Life Sciences. D.S.N. owns stock in Madrigal Pharmaceuticals, outside the submitted work. K.J.L. holds equity in Standard BioTools and is on the scientific advisory board for MBQ Pharma. S.A.S. reports equity in Agenus, Agios Pharmaceuticals, Breakbio, Bristol-Myers Squibb and Lumos Pharma. S.A.S. is a consultant for Imunon and Jivanu therapeutics. E.F.F. is an equity holder in and consultant for BioNTech, an equity holder and scientific advisory board member of BioEntre, and a founder and equity holder of Dionis Therapeutics. C.J.W. is subject to a conflict-of-interest management plan for the reported studies because of her former competing financial interests in Neon Therapeutics, which was acquired by BioNTech. Under this plan, C.J.W. may not access identifiable data for human participants or otherwise participate directly in the Institutional Review Board-approved protocol reported herein. C.J.W.’s contributions to the overall strategy and data analyses occurred on a de-identified basis. Patent applications have been filed on aspects of the described work entitled as follows: ‘Compositions and methods for personalized neoplasia vaccines’ (N.H., E.F.F. and C.J.W.), ‘Methods for identifying tumour specific neoantigens’ (N.H. and C.J.W.), ‘Formulations for neoplasia vaccines’ (E.F.F.) and ‘Combination therapy for neoantigen vaccine’ (N.H., C.J.W. and E.F.F.). The DFCI, the lead site of this trial, has a proprietary and financial interest in the personalized neoantigen vaccine. D.B.K. is a scientific advisor for Immunitrack, a wholly owned subsidiary of Eli Lilly and Company and Breakbio. D.B.K. owns equity in Affimed N.V., Agenus, Armata Pharmaceuticals, Breakbio, BioMarin Pharmaceuticals, Celldex Therapeutics, Editas Medicine, Immunitybio, Lexicon Pharmaceuticals, Summit Therapeutics and Viking Therapeutics. P.A.O. has received research funding from and/or has advised Agenus, Amgen, Armo BioSciences Array, AstraZeneca/MedImmune, Bristol-Meyers Squibb, Celldex, Compass Therapeutics, CytomX, Evaxion, Immunetune, Imunon, LGChem, Merck, Neon Therapeutics (now BioNTechUS), Novartis, Pharmajet, Phio, Pfizer, Oncorus, Roche/Genentech, Servier and Xencor. Committees: NCCN. T.K.C. reports grants, personal fees and non-financial support from Roche and Genentech during the conduct of the study; and reports institutional and/or personal, paid and/or unpaid support for research, advisory boards, consultancy, and/or honoraria from Alkermes, AstraZeneca, Aravive, Aveo, Bayer, Bristol Myers-Squibb, Calithera, Circle Pharma, Eisai, EMD Serono, Exelixis, GlaxoSmithKline, Gilead, IQVA, Infinity, Ipsen, Jansen, Kanaph, Lilly, Merck, Nikang, Nuscan, Novartis, Oncohost, Pfizer, Roche, Sanofi/Aventis, Scholar Rock, Surface Oncology, Takeda, Tempest, Up-To-Date, CME events (Peerview, OncLive, MJH and others), outside the submitted work. Institutional patents filed on molecular alterations and immunotherapy response/toxicity, and ctDNA. Equity: Tempest, Pionyr, Osel, Precede Bio, CureResponse, InnDura Therapeutics, Primium. Committees: NCCN, GU Steering Committee, ASCO/ESMO, ACCRU, KidneyCan, ODAC. Mentored several non-United States citizens on research projects with potential funding (in part) from non-United States sources/Foreign Components. The institution (Dana-Farber Cancer Institute) may have received additional independent funding of drug companies and/or royalties potentially involved in research around the subject matter. G.M., V.C., E.B., C.R.T., A.P.V., C.F., J.F., A.B.A., N.R.S., Y.L., S.L., J.S., S.L.C., M.S.H., O.O., A.M., H.G., C.B.P., M.M., I.C., A.T., J.D.-C., A.A.H., B.S., J.M.S., L.E., L.R.O., S.G. and G.O. have no reported disclosures related to the current work.
Figures











References
-
- Weber, J. S. et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): a randomised, phase 2b study. Lancet403, 632–644 (2024). - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- P50 CA101942/CA/NCI NIH HHS/United States
- K12 CA090354/CA/NCI NIH HHS/United States
- R37 CA279822/CA/NCI NIH HHS/United States
- R01 HL157174/HL/NHLBI NIH HHS/United States
- P01 CA206978/CA/NCI NIH HHS/United States
- R01 CA229261/CA/NCI NIH HHS/United States
- P30 CA006516/CA/NCI NIH HHS/United States
- U24 CA270823/CA/NCI NIH HHS/United States
- T32 CA009172/CA/NCI NIH HHS/United States
- P30 CA016359/CA/NCI NIH HHS/United States
- R50 CA251956/CA/NCI NIH HHS/United States
- T32 CA207021/CA/NCI NIH HHS/United States
- U01 CA271402/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
