

A Novel Missense Variant in SORBS2 Is Causative With Familial Alzheimer's Disease
- PMID: 39912518
- PMCID: PMC11800137
- DOI: 10.1111/cns.70256
A Novel Missense Variant in SORBS2 Is Causative With Familial Alzheimer's Disease
Abstract
Background: Alzheimer's disease (AD) is a common neurodegenerative disorder with a substantial genetic component. Despite advances in elucidating the genetic underpinnings of AD, much of its heritability remains unexplained. Discovering novel genetic variants and understanding their pathogenic roles are crucial challenges in AD research.
Objective: This study aimed to identify pathogenic genes and elucidate their role in familial early-onset AD (EOAD).
Methods: Blood samples from an EOAD pedigree and Sorbin and SH3 Domain-Containing Protein 2 (SORBS2) T189M transgenic mice were analyzed. Cognitive function was assessed via the Morris water maze (MWM). Protein expression was evaluated by western blotting, while amyloid-β (Aβ) levels were quantified via immunohistochemistry and enzyme-linked immunosorbent assay. Inflammatory markers were measured using immunofluorescence and quantitative reverse transcription polymerase chain reaction (PCR). Neuronal morphology, including dendritic and spine alterations, was examined using Golgi staining.
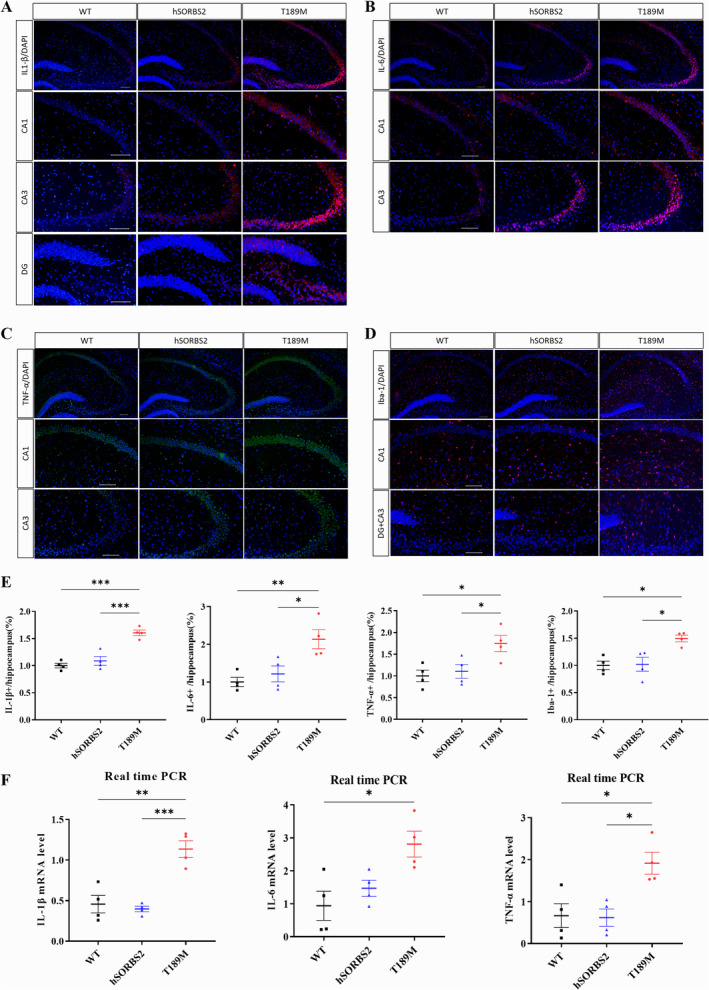
Results: We identified a novel SORBS2 variant (c. 566C>T, p. T189M) in a Han Chinese family, segregating with AD in a Mendelian fashion. SORBS2 T189M transgenic mice exhibited cognitive deficits, cortical Aβ accumulation, and an increased Aβ42/Aβ40 ratio. Additionally, elevated levels of interleukin (IL)-1β, IL-6, tumor necrosis factor α (TNF-α), and ionized calcium-binding adaptor molecule 1 (Iba1)-positive microglia, along with neuronal loss, were observed in the brains of T189M mice.
Conclusion: Our study suggest that the SORBS2 T189M variant is a novel candidate causal mutation associated with familial AD in a Chinese pedigree, contributing to AD pathogenesis by promoting neuroinflammation and neuronal injury. Notably, this study is the first to establish a link between SORBS2 mutations and AD.
Keywords: SORBS2; Alzheimer's disease; neuroinflammation; pathological mechanism.
© 2025 The Author(s). CNS Neuroscience & Therapeutics published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts interests.
Figures

 ) underwent whole genome sequencing. The proband is indicated by an arrow; black symbols represent affected members, white symbols represent unaffected members, squares indicate males, circles indicate females, and slashes denote deceased members. Age1/age2 in the top left corner of symbols indicates current age or age at death/age at onset. (+) indicates mutation carriers, and (−) indicates non‐carriers. NA indicates data not available. (B)11C‐PIB PET imaging of the proband confirms amyloid deposition, consistent with AD pathology. (C) Sequencing chromatogram showing the heterozygous SORBS2 c.566C>T mutation in affected family members.
) underwent whole genome sequencing. The proband is indicated by an arrow; black symbols represent affected members, white symbols represent unaffected members, squares indicate males, circles indicate females, and slashes denote deceased members. Age1/age2 in the top left corner of symbols indicates current age or age at death/age at onset. (+) indicates mutation carriers, and (−) indicates non‐carriers. NA indicates data not available. (B)11C‐PIB PET imaging of the proband confirms amyloid deposition, consistent with AD pathology. (C) Sequencing chromatogram showing the heterozygous SORBS2 c.566C>T mutation in affected family members.


References
MeSH terms
Substances
Supplementary concepts
Grants and funding
- 2021ZD0201803/the STI2030-Major Projects
- 2021ZD0201802/the STI2030-Major Projects
- 81530036/the Key Project of the National Natural Science Foundation of China
- U20A20354/the Key Project of the National Natural Science Foundation of China
- Z201100005520016/Beijing Brain Initiative from Beijing Municipal Science & Technology Commission
LinkOut - more resources
Full Text Sources
Medical
