

R-loops acted on by RNase H1 influence DNA replication timing and genome stability in Leishmania
- PMID: 39922816
- PMCID: PMC11807225
- DOI: 10.1038/s41467-025-56785-y
R-loops acted on by RNase H1 influence DNA replication timing and genome stability in Leishmania
Abstract
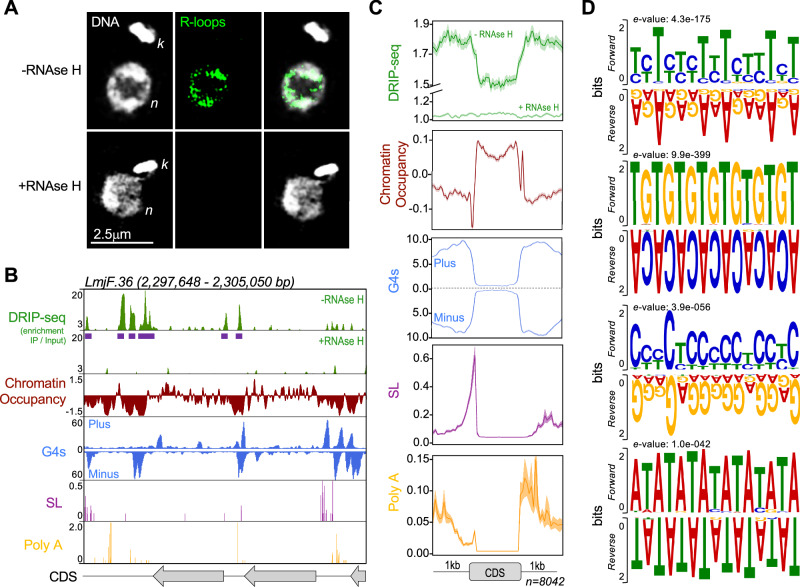
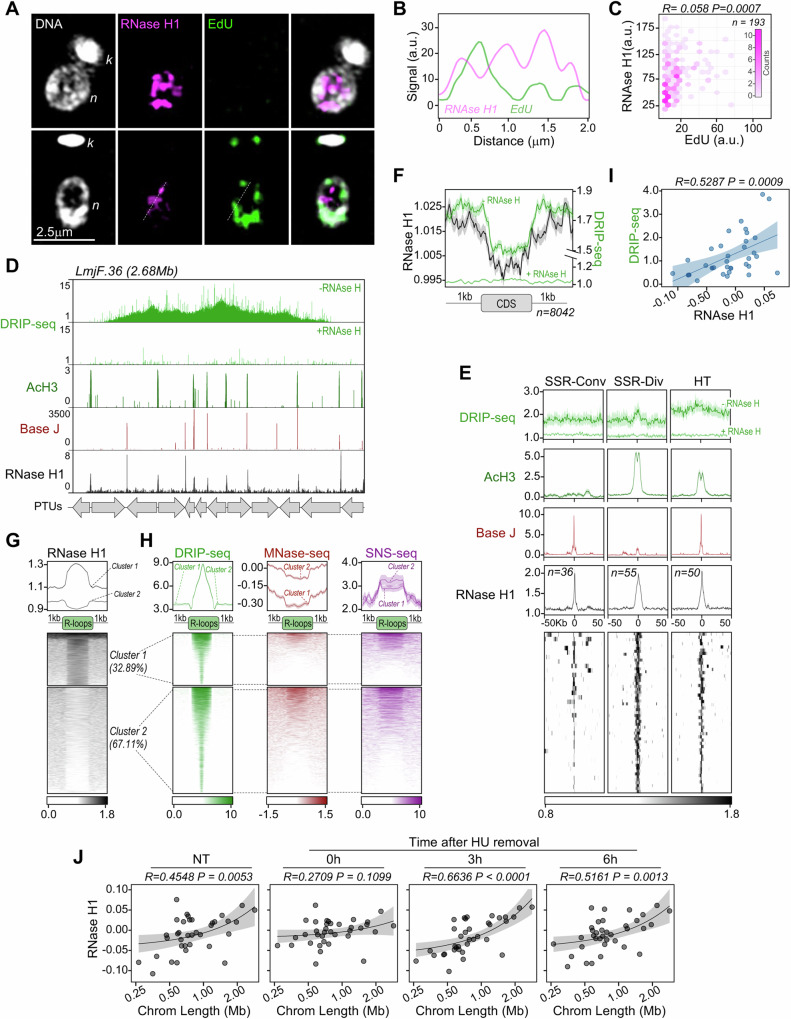
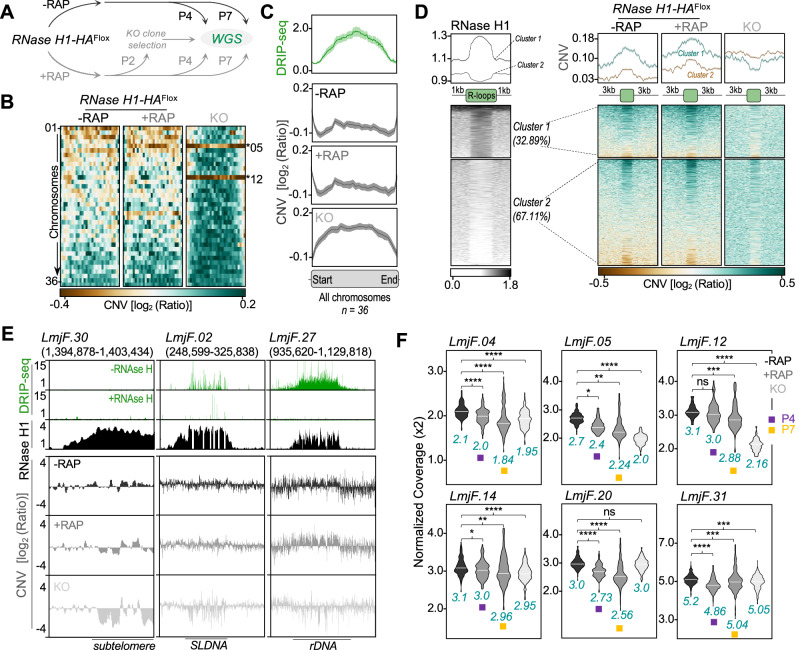
Genomes in eukaryotes normally undergo DNA replication in a choreographed temporal order, resulting in early and late replicating chromosome compartments. Leishmania, a human protozoan parasite, displays an unconventional DNA replication program in which the timing of DNA replication completion is chromosome size-dependent: larger chromosomes complete replication later then smaller ones. Here we show that both R-loops and RNase H1, a ribonuclease that resolves RNA-DNA hybrids, accumulate in Leishmania major chromosomes in a pattern that reflects their replication timing. Furthermore, we demonstrate that such differential organisation of R-loops, RNase H1 and DNA replication timing across the parasite's chromosomes correlates with size-dependent differences in chromatin accessibility, G quadruplex distribution and sequence content. Using conditional gene excision, we show that loss of RNase H1 leads to transient growth perturbation and permanently abrogates the differences in DNA replication timing across chromosomes, as well as altering levels of aneuploidy and increasing chromosome instability in a size-dependent manner. This work provides a link between R-loop homeostasis and DNA replication timing in a eukaryotic parasite and demonstrates that orchestration of DNA replication dictates levels of genome plasticity in Leishmania.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures



References
MeSH terms
Substances
Grants and funding
- BB/W001101/1/RCUK | Biotechnology and Biological Sciences Research Council (BBSRC)
- 224501/Z/21/Z/Wellcome Trust (Wellcome)
- MR/S019472/1/RCUK | Medical Research Council (MRC)
- RECREPEMLE/EC | Horizon 2020 Framework Programme (EU Framework Programme for Research and Innovation H2020)
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
