

ISG15-Dependent Stabilisation of USP18 Is Necessary but Not Sufficient to Regulate Type I Interferon Signalling in Humans
- PMID: 39931755
- PMCID: PMC11811815
- DOI: 10.1002/eji.202451651
ISG15-Dependent Stabilisation of USP18 Is Necessary but Not Sufficient to Regulate Type I Interferon Signalling in Humans
Abstract
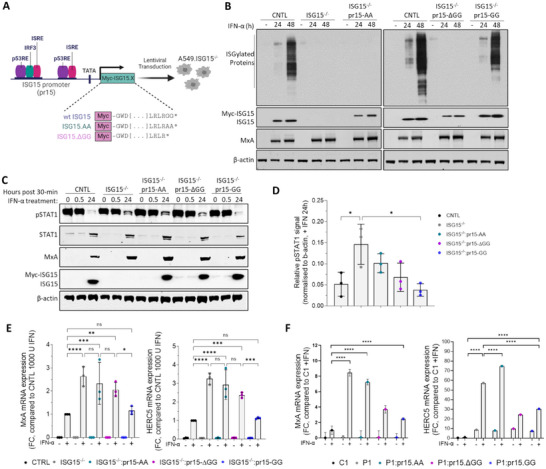
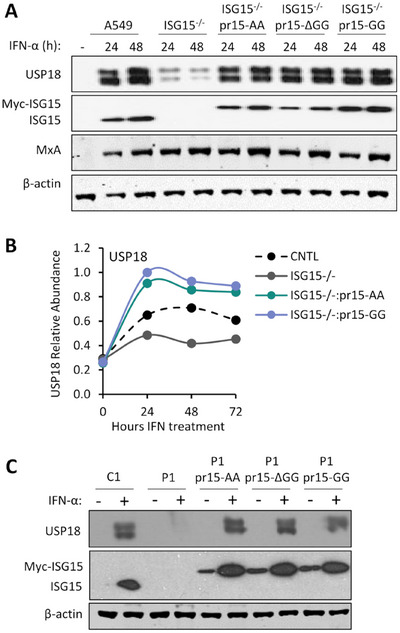
Type I interferon (IFN) signalling induces the expression of several hundred IFN-stimulated genes (ISGs) that provide an unfavourable environment for viral replication. To prevent an overexuberant response and autoinflammatory disease, IFN signalling requires tight control. One critical regulator is the ubiquitin-like protein IFN-stimulated gene 15 (ISG15), evidenced by autoinflammatory disease in patients with inherited ISG15 deficiencies. Current models suggest that ISG15 stabilises ubiquitin-specific peptidase 18 (USP18), a well-established negative regulator of IFN signalling. USP18 also functions as an ISG15-specific peptidase that cleaves ISG15 from ISGylated proteins; however, USP18's catalytic activity is dispensable for controlling IFN signalling. Here, we show that the ISG15-dependent stabilisation of USP18 involves hydrophobic interactions reliant on tryptophan 123 (W123) in ISG15. Nonetheless, while USP18 stabilisation is necessary, it is not sufficient for the regulation of IFN signalling; ISG15 C-terminal mutants with significantly reduced affinity still stabilised USP18, yet the magnitude of signalling resembled ISG15-deficient cells. Hence, USP18 requires non-covalent interactions with the ISG15 C-terminal diGlycine motif to promote its regulatory function. It shows ISG15 is a repressor of type I IFN signalling beyond its role as a USP18 stabiliser.
Keywords: ISG15; Type I IFN signalling; USP18; autoinflammatory disease; interferonopathy; interferon‐stimulated genes.
© 2025 The Author(s). European Journal of Immunology published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures





References
-
- Gauzzi M. C., Velazquez L., McKendry R., Mogensen K. E., Fellous M., and Pellegrini S., “Interferon‐alpha‐dependent Activation of Tyk2 Requires Phosphorylation of Positive Regulatory Tyrosines by another Kinase,” Journal of Biological Chemistry 271 (1996): 20494–20500, 10.1074/jbc.271.34.20494. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
