

SOD1-related inherited peripheral neuropathies in a Japanese cohort: genetic variants and clinical insights
- PMID: 39932579
- PMCID: PMC11814053
- DOI: 10.1007/s00415-025-12925-4
SOD1-related inherited peripheral neuropathies in a Japanese cohort: genetic variants and clinical insights
Abstract
Background: Inherited peripheral neuropathies (IPNs) encompass a wide range of disorders affecting the peripheral nervous system, often with complex genetic causes and frequent underdiagnosis. The variants in the superoxide dismutase 1 (SOD1) gene, primarily linked to amyotrophic lateral sclerosis (ALS), have also been associated with peripheral neuropathy. The recent approval of Tofersen, targeting SOD1-related ALS, highlights the importance of precise genetic diagnosis. This study explores the clinical and genetic profiles of SOD1-related IPNs (SOD1-IPN) in a nationwide Japanese IPN cohort.
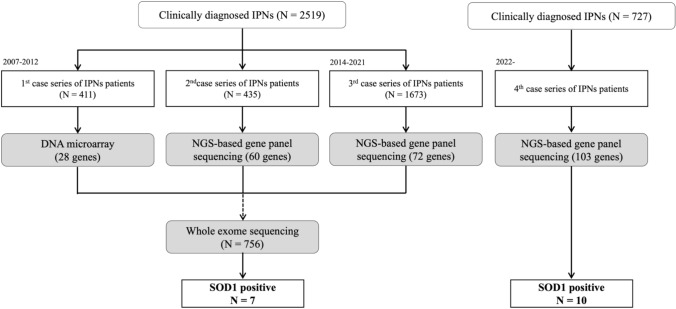
Methods: Clinical and genetic data were assessed from 1483 Japanese patients with IPN, with a focus on those harboring SOD1 pathogenic variants. The clinical evaluations included age of onset, gender, muscle weakness patterns, sensory disturbances, reflex responses, and electrophysiological findings.
Results: Seventeen patients with SOD1 pathogenic variants were identified, reinforcing SOD1's role in IPN. The average onset age was 47, with a slight male predominance. Distal muscle weakness was noted in 9 of 13 patients, and asymmetric muscle weakness and atrophy in 10 of 14 cases. Mild sensory disturbances were observed in eight patients, with some showing hyperreflexia and abnormal reflexes. Electrophysiology predominantly indicated a length-dependent, motor-dominant axonal neuropathy.
Conclusion: This study reveals the clinical variability and likely underdiagnosis of SOD1-IPN, supporting the integration of SOD1 screening in IPN genetic testing, especially for patients with asymmetric, length-dependent axonal neuropathy evident in clinical and electrophysiological assessments.
Keywords: Amyotrophic lateral sclerosis; Electrophysiology; Inherited peripheral neuropathy; SOD1.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Conflicts of interest: The authors report that there is no conflicts of interests.
Figures
References
-
- Higuchi Y, Takashima H (2023) Clinical genetics of Charcot-Marie-Tooth disease. J Hum Genet 68(3):199–214. 10.1038/s10038-022-01031-2 - PubMed
-
- Østern R, Fagerheim T, Ørstavik K et al (2012) Hereditary motor neuron disease in a large Norwegian family with a “H46R” substitution in the superoxide dismutase 1 gene. Neuromuscul Disord 22(6):511–521. 10.1016/j.nmd.2012.01.011 - PubMed
-
- Querin G, Corcia P, Lenglet T et al (2017) Motor neuron disease of very long disease duration or Charcot-Marie-Tooth disease? A novel phenotype related to the SOD1 p.E22G variant. Rev Neurol (Paris) 173(10):671–673. 10.1016/j.neurol.2017.05.008 - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
