

Accelerated epigenetic aging in Huntington's disease involves polycomb repressive complex 1
- PMID: 39934111
- PMCID: PMC11814324
- DOI: 10.1038/s41467-025-56722-z
Accelerated epigenetic aging in Huntington's disease involves polycomb repressive complex 1
Abstract
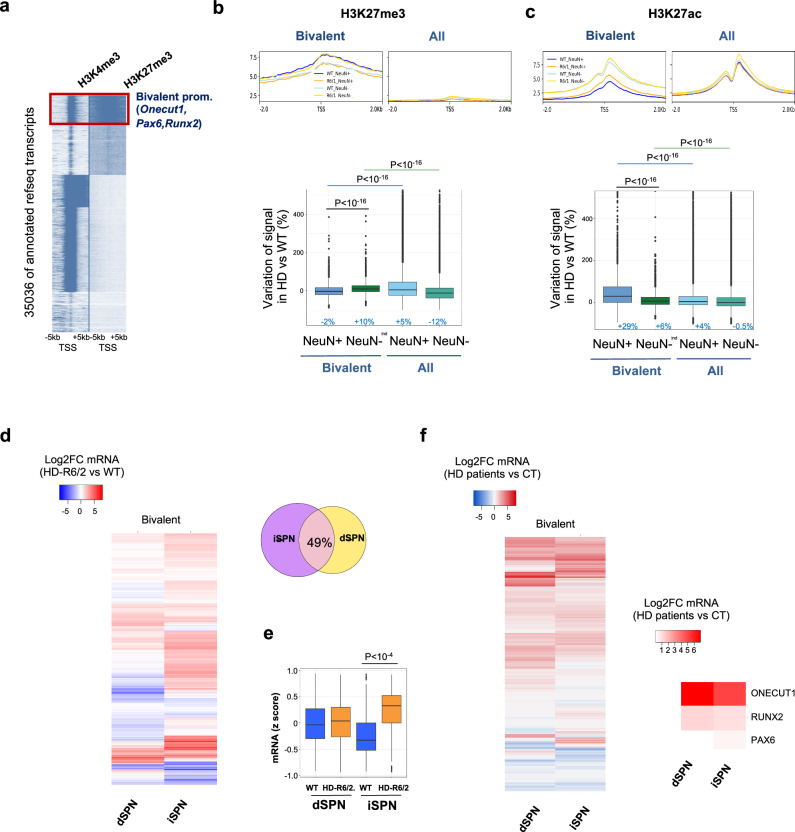
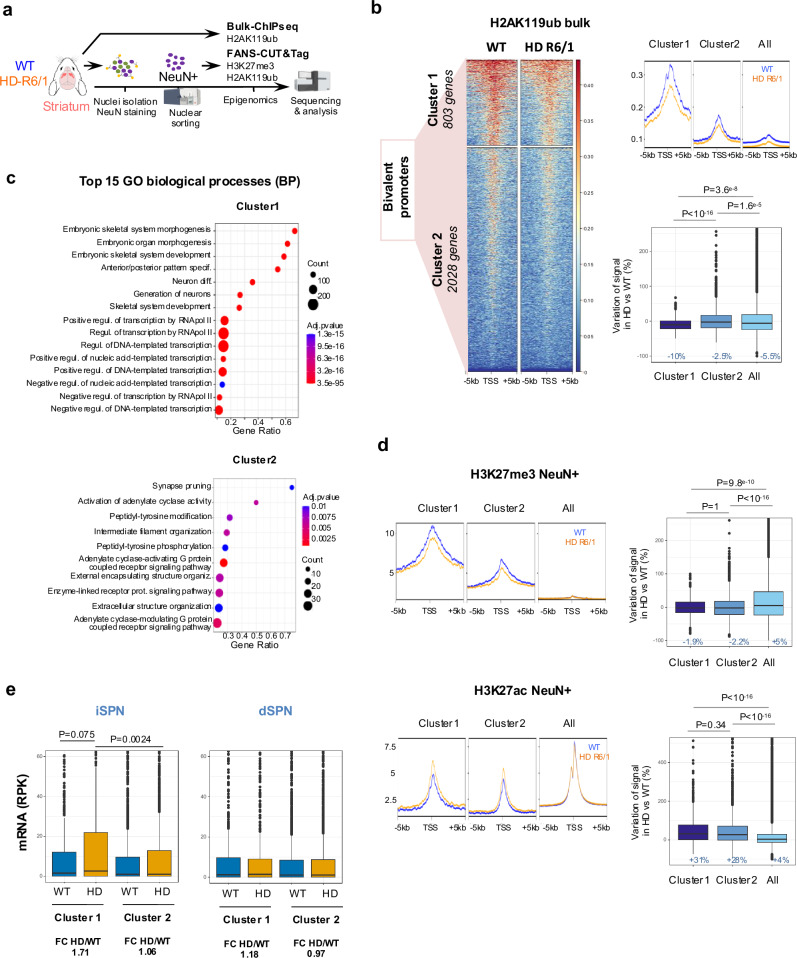
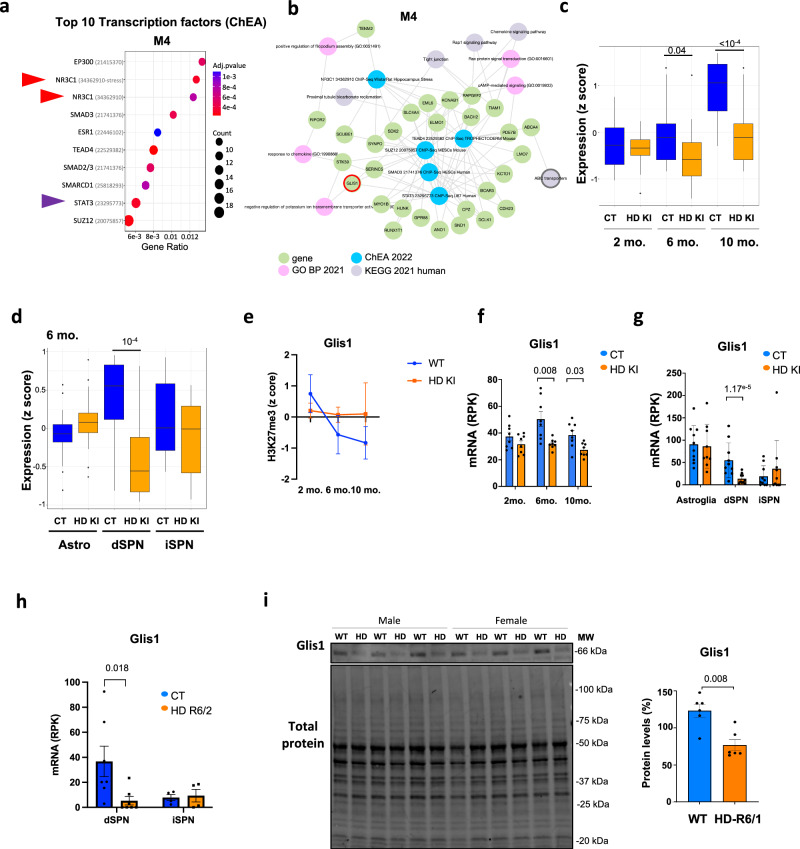
Loss of epigenetic information during physiological aging compromises cellular identity, leading to de-repression of developmental genes. Here, we assessed the epigenomic landscape of vulnerable neurons in two reference mouse models of Huntington neurodegenerative disease (HD), using cell-type-specific multi-omics, including temporal analysis at three disease stages via FANS-CUT&Tag. We show accelerated de-repression of developmental genes in HD striatal neurons, involving histone re-acetylation and depletion of H2AK119 ubiquitination and H3K27 trimethylation marks, which are catalyzed by polycomb repressive complexes 1 and 2 (PRC1 and PRC2), respectively. We further identify a PRC1-dependent subcluster of bivalent developmental transcription factors that is re-activated in HD striatal neurons. This mechanism likely involves progressive paralog switching between PRC1-CBX genes, which promotes the upregulation of normally low-expressed PRC1-CBX2/4/8 isoforms in striatal neurons, alongside the down-regulation of predominant PRC1-CBX isoforms in these cells (e.g., CBX6/7). Collectively, our data provide evidence for PRC1-dependent accelerated epigenetic aging in HD vulnerable neurons.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures




References
-
- Bates, G. P. et al. Huntington disease. Nat. Rev. Dis. Prim.1, 15005 (2015). - PubMed
-
- Vonsattel, J. P., Keller, C. & Cortes Ramirez, E. P. Huntington’s disease - neuropathology. Handb. Clin. Neurol.100, 83–100 (2011). - PubMed
-
- Heinsen, H. et al. Cortical and striatal neurone number in Huntington’s disease. Acta Neuropathol.88, 320–333 (1994). - PubMed
-
- Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell186, 243–278 (2023). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
