

Progress in Research on Regulated Cell Death in Cerebral Ischaemic Injury After Cardiac Arrest
- PMID: 39936900
- PMCID: PMC11816164
- DOI: 10.1111/jcmm.70404
Progress in Research on Regulated Cell Death in Cerebral Ischaemic Injury After Cardiac Arrest
Abstract
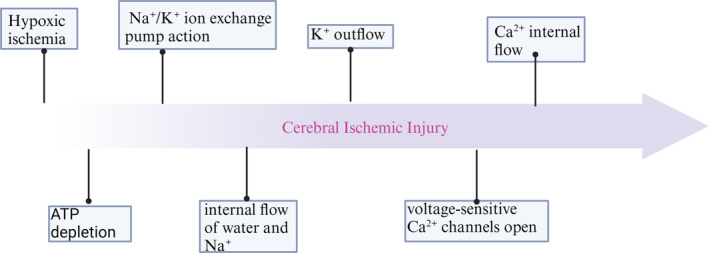
Ischaemic damage to the brain is the main cause of brain injury after cardiac arrest. The current treatment focuses on early reperfusion, but reperfusion tends to cause reperfusion injury, which is a significant problem. Cell death is an irreversible and normal end to cell life, playing key roles in maintaining the homeostasis and development of multicellular organisms. To date, cell death can be classified into two categories: accidental cell death (ACD) and regulated cell death (RCD). Cell death plays an indispensable role in cerebral ischaemia injury. An increasing number of scholars are exploring the mechanisms and sites of cell death during targeted inhibition of cerebral ischaemia to treat cerebral ischaemia injury. In addition to the established cell death pathways, namely, the apoptosis, pyroptosis and necroptosis pathways, ferroptosis and cuproptosis pathways have been discovered. This article reviews the cell death pathways involved in ischaemic brain injury, discusses the roles played by these death modalities, and suggests therapeutic directions for future targeting of cell death sites.
Keywords: cerebral ischaemic injury; cuproptosis; mechanism; regulated cell death; therapy.
© 2025 The Author(s). Journal of Cellular and Molecular Medicine published by Foundation for Cellular and Molecular Medicine and John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures



Similar articles
-
Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research.J Hematol Oncol. 2022 Dec 8;15(1):174. doi: 10.1186/s13045-022-01392-3. J Hematol Oncol. 2022. PMID: 36482419 Free PMC article. Review.
-
[The role of regulated cell death in renal ischemia-reperfusion injury].Sheng Li Xue Bao. 2022 Apr 25;74(2):320-332. Sheng Li Xue Bao. 2022. PMID: 35503080 Review. Chinese.
-
Studies of isolated global brain ischaemia: I. Overview of irreversible brain injury and evolution of a new concept - redefining the time of brain death.Eur J Cardiothorac Surg. 2012 May;41(5):1132-7. doi: 10.1093/ejcts/ezr315. Epub 2012 Mar 6. Eur J Cardiothorac Surg. 2012. PMID: 22398465 Review.
-
Inhibition of autophagy-dependent pyroptosis attenuates cerebral ischaemia/reperfusion injury.J Cell Mol Med. 2021 Jun;25(11):5060-5069. doi: 10.1111/jcmm.16483. Epub 2021 May 2. J Cell Mol Med. 2021. PMID: 33938129 Free PMC article.
-
Mechanisms and Therapeutic Potential of Multiple Forms of Cell Death in Myocardial Ischemia-Reperfusion Injury.Int J Mol Sci. 2024 Dec 17;25(24):13492. doi: 10.3390/ijms252413492. Int J Mol Sci. 2024. PMID: 39769255 Free PMC article. Review.
Cited by
-
Factors Contributing to Resistance to Ischemia-Reperfusion Injury in Olfactory Mitral Cells.Int J Mol Sci. 2025 May 25;26(11):5079. doi: 10.3390/ijms26115079. Int J Mol Sci. 2025. PMID: 40507890 Free PMC article. Review.
References
-
- Sasson C., Rogers M. A., Dahl J., and Kellermann A. L., “Predictors of Survival From Out‐Of‐Hospital Cardiac Arrest: A Systematic Review and Meta‐Analysis,” Circulation. Cardiovascular Quality and Outcomes 3, no. 1 (2010): 63–81. - PubMed
-
- Myat A., Song K. J., and Rea T., “Out‐Of‐Hospital Cardiac Arrest: Current Concepts,” Lancet 391, no. 10124 (2018): 970–979. - PubMed
-
- Imberti R., Bellinzona G., Riccardi F., Pagani M., and Langer M., “Cerebral Perfusion Pressure and Cerebral Tissue Oxygen Tension in a Patient During Cardiopulmonary Resuscitation,” Intensive Care Medicine 29, no. 6 (2003): 1016–1019. - PubMed
-
- Nolan J. P., Neumar R. W., Adrie C., et al., “Post‐Cardiac Arrest Syndrome: Epidemiology, Pathophysiology, Treatment, and Prognostication. A Scientific Statement From the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke,” Resuscitation 79, no. 3 (2008): 350–379. - PubMed
-
- Soleimanpour H., Khoshnudi F., Movaghar M. H., and Ziapour B., “Improvement of Decerebrate Status in a Hanged Child Following Emergent Tracheostomy,” Pakistan Journal of Biological Sciences 13, no. 23 (2010): 1164–1165. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
