

Combined genomics and proteomics unveils elusive variants and vast aetiologic heterogeneity in dystonia
- PMID: 39937650
- PMCID: PMC12316014
- DOI: 10.1093/brain/awaf059
Combined genomics and proteomics unveils elusive variants and vast aetiologic heterogeneity in dystonia
Abstract
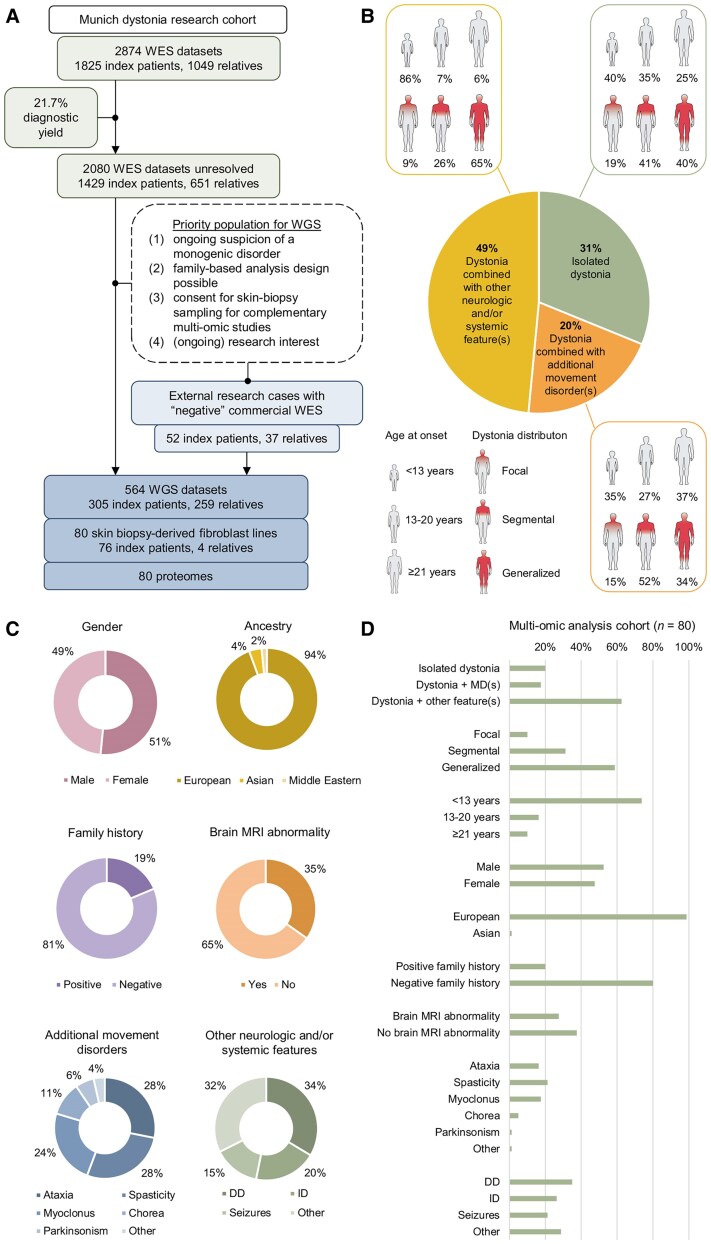
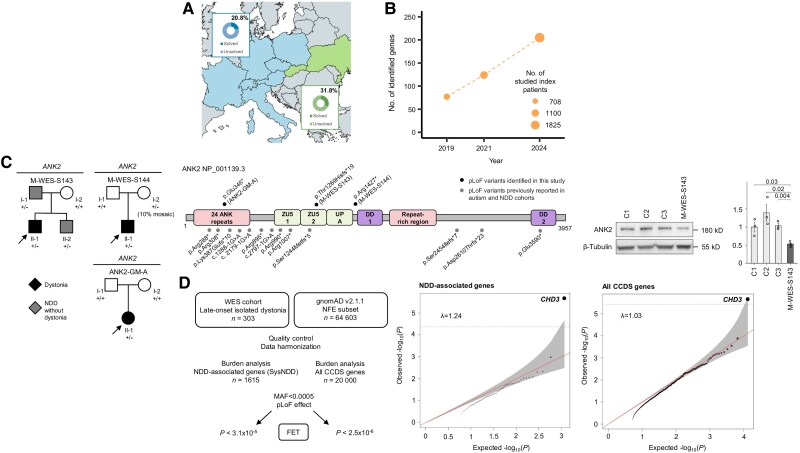
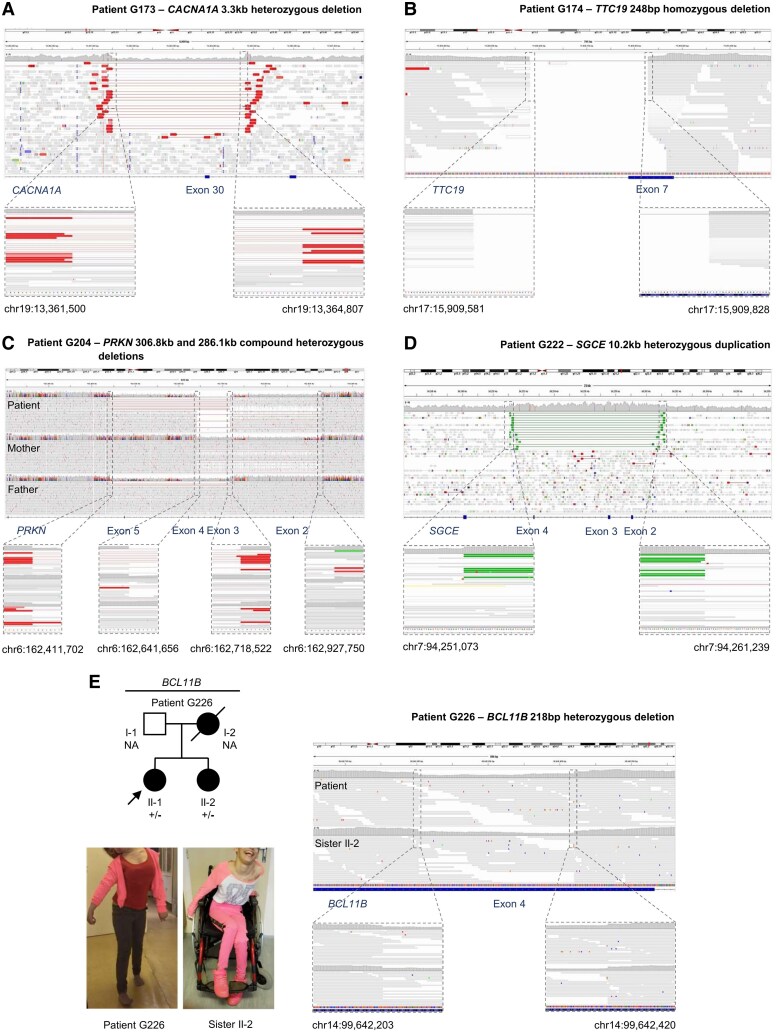
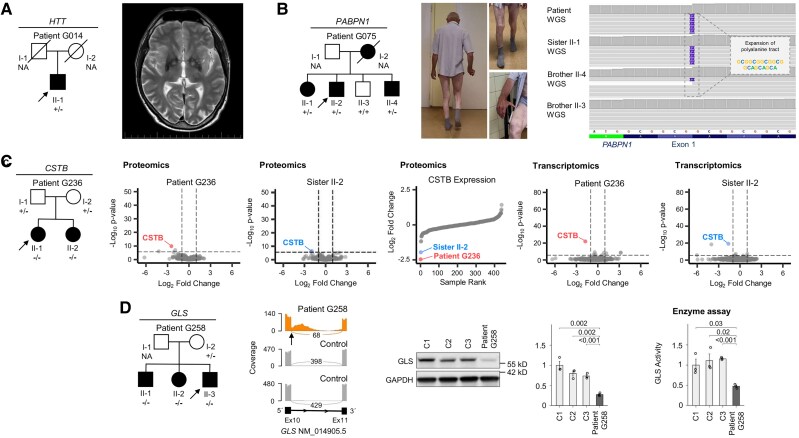
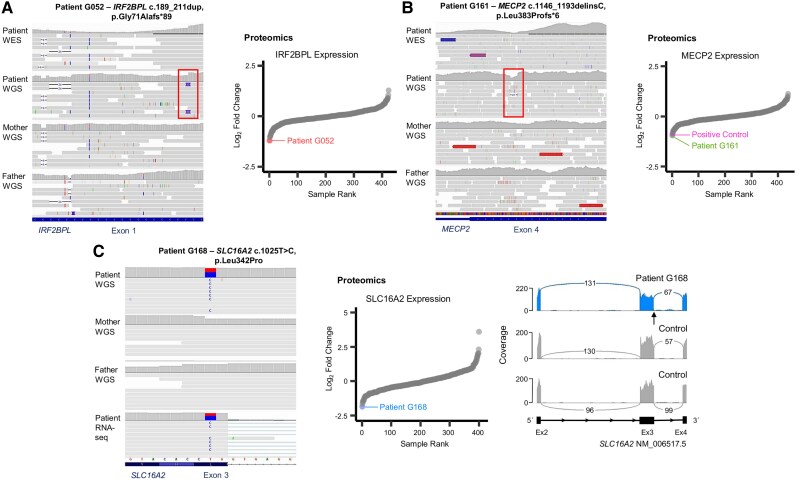
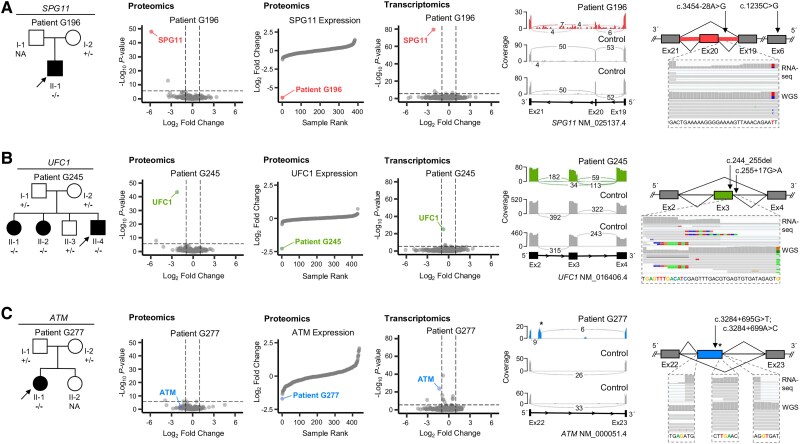
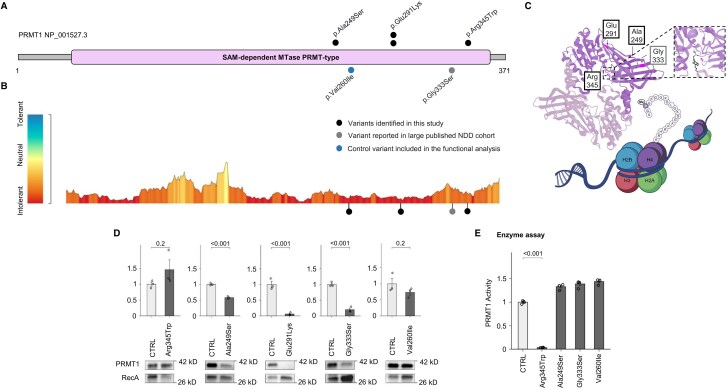
Dystonia is a rare disease trait for which large-scale genomic investigations are still underrepresented. Genetic heterogeneity among patients with unexplained dystonia warrants interrogation of entire genome sequences, but this has not yet been systematically evaluated. To significantly enhance our understanding of the genetic contribution to dystonia, we (re)analysed 2874 whole-exome sequencing (WES), 564 whole-genome sequencing (WGS), as well as 80 fibroblast-derived proteomics datasets, representing the output of high-throughput analyses in 1990 patients and 973 unaffected relatives from 1877 families. Recruitment and precision-phenotyping procedures were driven by long-term collaborations of international experts with access to overlooked populations. By exploring WES data, we found that continuous scaling of sample sizes resulted in steady gains in the number of associated disease genes without plateauing. On average, every second diagnosis involved a gene not previously implicated in our cohort. Second-line WGS focused on a subcohort of undiagnosed individuals with high likelihood of having monogenic forms of dystonia, comprising large proportions of patients with early onset (81.3%), generalized symptom distribution (50.8%) and/or coexisting features (68.9%). We undertook extensive searches for variants in nuclear and mitochondrial genomes to uncover 38 (ultra)rare diagnostic-grade findings in 37 of 305 index patients (12.1%), many of which had remained undetected due to methodological inferiority of WES or pipeline limitations. WGS-identified elusive variations included alterations in exons poorly covered by WES, RNA-gene variants, mitochondrial-DNA mutations, small copy-number variants, complex rearranged genome structure and short tandem repeats. For improved variant interpretation in WGS-inconclusive cases, we employed systematic integration of quantitative proteomics. This aided in verifying diagnoses related to technically challenging variants and in upgrading a variant of uncertain significance (3 of 70 WGS-inconclusive index patients, 4.3%). Further, unsupervised proteomic outlier analysis supplemented with transcriptome sequencing revealed pathological gene underexpression induced by transcript disruptions in three more index patients with underlying (deep) intronic variants (3/70, 4.3%), highlighting the potential for targeted antisense-oligonucleotide therapy development. Finally, trio-WGS prioritized a de novo missense change in the candidate PRMT1, encoding a histone methyltransferase. Data-sharing strategies supported the discovery of three distinct PRMT1 de novo variants in four phenotypically similar patients, associated with loss-of-function effects in in vitro assays. This work underscores the importance of continually expanding sequencing cohorts to characterize the extensive spectrum of gene aberrations in dystonia. We show that a pool of unresolved cases is amenable to WGS and complementary multi-omic studies, directing advanced aetiopathological concepts and future diagnostic-practice workflows for dystonia.
Keywords: dystonia; genomics; multi-omics; proteomics; transcriptomics; whole-genome sequencing.
© The Author(s) 2025. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors report no competing interests.
Figures
Similar articles
-
Can a Liquid Biopsy Detect Circulating Tumor DNA With Low-passage Whole-genome Sequencing in Patients With a Sarcoma? A Pilot Evaluation.Clin Orthop Relat Res. 2025 Jan 1;483(1):39-48. doi: 10.1097/CORR.0000000000003161. Epub 2024 Jun 21. Clin Orthop Relat Res. 2025. PMID: 38905450
-
Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19.Cochrane Database Syst Rev. 2022 May 20;5(5):CD013665. doi: 10.1002/14651858.CD013665.pub3. Cochrane Database Syst Rev. 2022. PMID: 35593186 Free PMC article.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2021 Apr 19;4(4):CD011535. doi: 10.1002/14651858.CD011535.pub4. Cochrane Database Syst Rev. 2021. Update in: Cochrane Database Syst Rev. 2022 May 23;5:CD011535. doi: 10.1002/14651858.CD011535.pub5. PMID: 33871055 Free PMC article. Updated.
-
Genetic Diversity and Expanded Phenotypes in Dystonia: Insights from Large-Scale Exome Sequencing.medRxiv [Preprint]. 2024 Dec 5:2024.12.02.24316741. doi: 10.1101/2024.12.02.24316741. medRxiv. 2024. Update in: Ann Clin Transl Neurol. 2025 Aug;12(8):1648-1659. doi: 10.1002/acn3.70100. PMID: 39677454 Free PMC article. Updated. Preprint.
-
Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis.Cochrane Database Syst Rev. 2020 Jan 9;1(1):CD011535. doi: 10.1002/14651858.CD011535.pub3. Cochrane Database Syst Rev. 2020. Update in: Cochrane Database Syst Rev. 2021 Apr 19;4:CD011535. doi: 10.1002/14651858.CD011535.pub4. PMID: 31917873 Free PMC article. Updated.
Cited by
-
Decoding the Alphabet Soup: A Practical Guide to Genetic Testing in Hyperkinetic Movement Disorders.Tremor Other Hyperkinet Mov (N Y). 2025 Jun 26;15:28. doi: 10.5334/tohm.971. eCollection 2025. Tremor Other Hyperkinet Mov (N Y). 2025. PMID: 40584247 Free PMC article.
-
Insights on the Shared Genetic Landscape of Neurodevelopmental and Movement Disorders.Curr Neurol Neurosci Rep. 2025 Mar 17;25(1):24. doi: 10.1007/s11910-025-01414-w. Curr Neurol Neurosci Rep. 2025. PMID: 40095113 Free PMC article. Review.
-
Expanding the Allelic and Clinical Heterogeneity of Movement Disorders Linked to Defects of Mitochondrial Adenosine Triphosphate Synthase.Mov Disord. 2025 Jul;40(7):1388-1400. doi: 10.1002/mds.30209. Epub 2025 Apr 25. Mov Disord. 2025. PMID: 40276935 Free PMC article.
References
-
- van Egmond ME, Kuiper A, Eggink H, et al. Dystonia in children and adolescents: A systematic review and a new diagnostic algorithm. J Neurol Neurosurg Psychiatry. 2015;86:774–781. - PubMed
-
- van Egmond ME, Lagrand TJ, Lizaitiene G, Smit M, Tijssen MAJ. A novel diagnostic approach for patients with adult-onset dystonia. J Neurol Neurosurg Psychiatry. 2022;93:1039–1048. - PubMed
MeSH terms
Grants and funding
- #458949627/DFG Research Infrastructure NGS_CC
- 423957469/DFG Research Infrastructure NGS_CC
- ZE 1213/2-1/DFG Research Infrastructure NGS_CC
- SCHO 1644/4-1/DFG Research Infrastructure NGS_CC
- WI 1820/14-1/DFG Research Infrastructure NGS_CC
- #458949627/DFG Research Infrastructure NGS_CC
- 01GM2302/Federal Ministry of Education and Research
- 2022_EKSE.185/Federal Ministry of Education and Research
- LX22NPO5107/National Institute for Neurological Research
- European Union
- NW24-04-0006/Next Generation EU
- APVV-22-0279/Next Generation EU
- 09I03-03-V03-00007/Next Generation EU
- 1/0712/22/Next Generation EU
- GR-2009-1594645/Next Generation EU
- 825575/Fondazione Regionale per la Ricerca Biomedica
- 01GM1906A/Fondazione Regionale per la Ricerca Biomedica
LinkOut - more resources
Full Text Sources
Medical
