

A1AT dysregulation of metabolically stressed hepatocytes by Kupffer cells drives MASH and fibrosis
- PMID: 39939782
- PMCID: PMC11873038
- DOI: 10.1038/s12276-025-01408-1
A1AT dysregulation of metabolically stressed hepatocytes by Kupffer cells drives MASH and fibrosis
Abstract
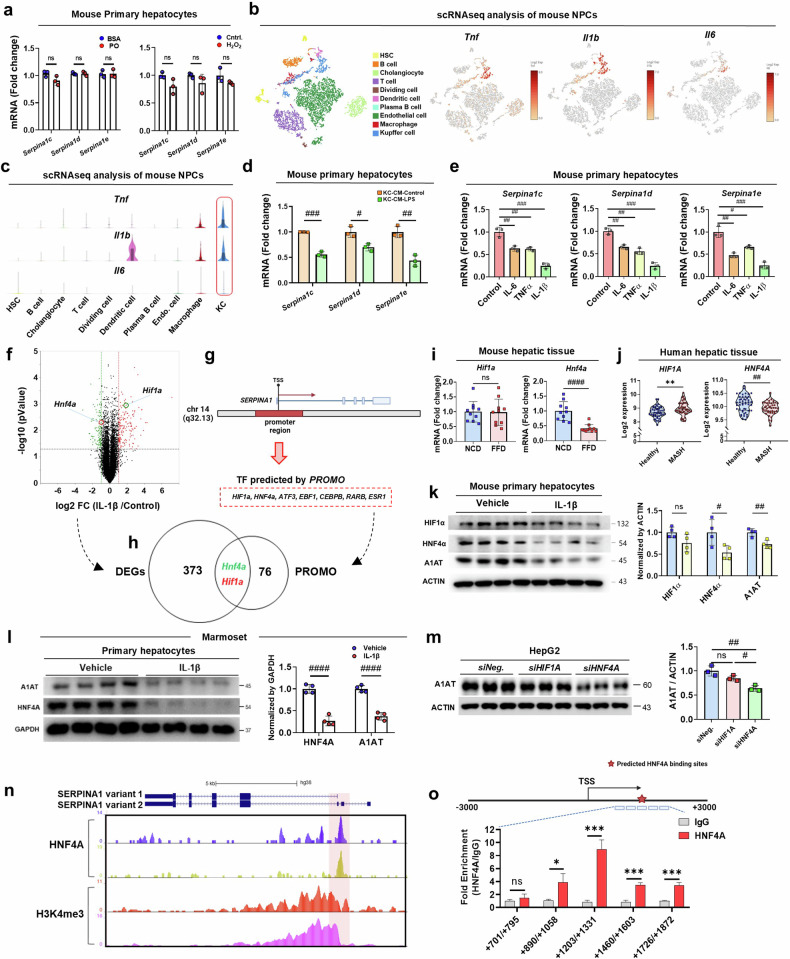
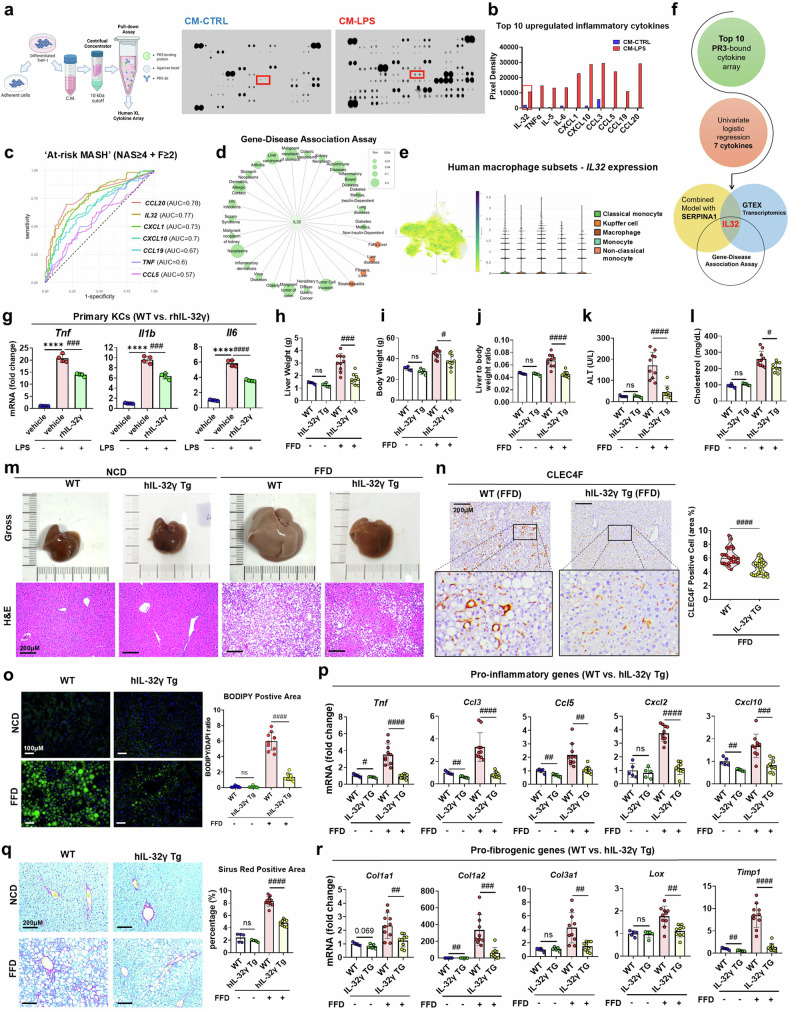
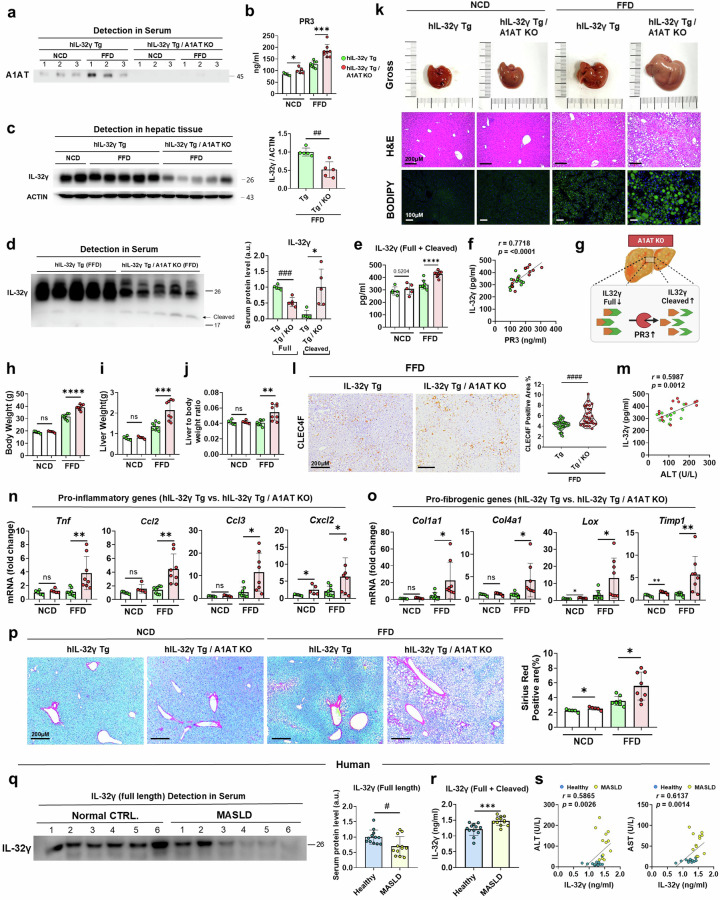
Metabolic dysfunction-associated steatohepatitis (MASH) is associated with the activation of Kupffer cells (KCs) and hepatic stellate cells, at which point a metabolically stressed hepatocyte becomes integral to the progression of the disease. We observed a significant reduction in the level of alpha-1-antitrypsin (A1AT), a hepatocyte-derived secreted factor, in both patients with MASH and mice fed a fast-food diet (FFD). KC-mediated hepatic inflammation, most notably IL-1β, led to the transcriptional inhibition of A1AT by HNF4α. In quintuple Serpina1a-e knockout mice, ablation of A1AT worsened MASH through increased activity of proteinase 3 (PR3), a proinflammatory protease produced by F4/80hi/CD11blow/TIM4-/CCR2+ monocyte-derived KCs (MoKCs). Conversely, A1AT restoration or PR3 inhibition mitigated MASH progression. A PR3-bound cytokine array identified IL-32 as a key factor associated with MASH. Combining IL-32 with SERPINA1, the gene encoding A1AT, synergistically predicted patients at risk of MASH through univariate logistic regression analysis. Furthermore, in vivo overexpression of IL-32γ alleviated MASH induced by FFD. However, additional knockout of A1AT increased PR3 activity, consequently abolishing the anti-MASH effects of IL-32γ. Blocking PR3-mediated IL-32γ cleavage via the V104A mutation sustained its protective actions, while the PR3-cleaved C-terminal fragment activated KCs. Additionally, after cleavage, the antifibrogenic effect of IL-32γ is lost, resulting in a failure to prevent the activation of hepatic stellate cells. This study highlights the critical role of hepatocyte-derived A1AT in the PR3/IL-32γ axis during MASH development. Strategies to correct A1AT dysregulation, such as A1AT supplementation or PR3 inhibition with sivelestat, may offer protection against the development and progression of MASH and fibrosis. Elevated hepatic IL-1β levels in MASH lead to the downregulation of A1AT via the transcription factor HNF4α, resulting in increased recruitment of proinflammatory MoKCs and heightened PR3 activity. PR3 cleaves IL-32γ, transforming it from an anti-inflammatory and antifibrogenic cytokine into a potent activator of KCs and failing to prevent HSC activation. This cascade amplifies liver inflammation and fibrosis, suggesting that targeting the A1AT/PR3/IL-32γ axis could be a strategy for treating MASH.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures






References
-
- Jezela-Stanek, A. & Chorostowska-Wynimko, J. Beyond the lungs: alpha-1 antitrypsin’s potential role in human gestation. Adv. Clin. Exp. Med. 28, 1257–1261 (2019). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
