

Antisense oligonucleotides modulate aberrant inclusion of poison exons in SCN1A-related Dravet syndrome
- PMID: 39946203
- PMCID: PMC11981616
- DOI: 10.1172/jci.insight.188014
Antisense oligonucleotides modulate aberrant inclusion of poison exons in SCN1A-related Dravet syndrome
Abstract
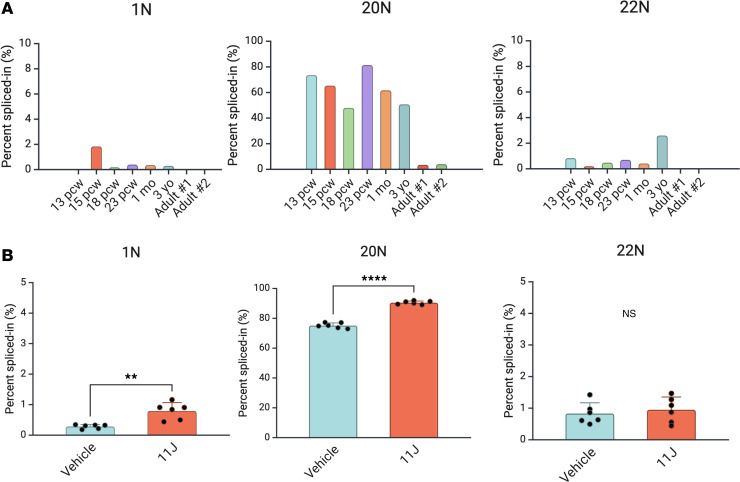
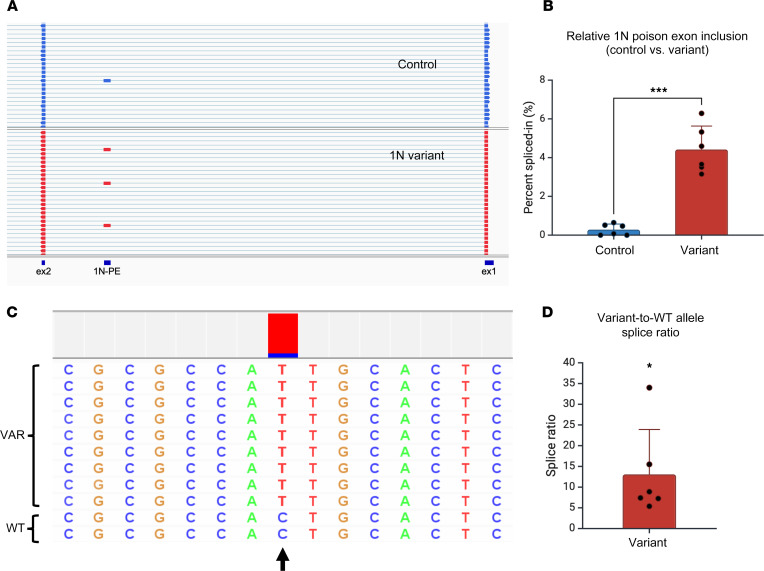
Dravet syndrome is a developmental and epileptic encephalopathy associated with pathogenic variants in SCN1A. Most disease-causing variants are located within coding regions, but recent work has shed light on the role of noncoding variants associated with a poison exon in intron 20 of SCN1A. Discovery of the SCN1A poison exon known as 20N has led to the first potential disease-modifying therapy for Dravet syndrome in the form of an antisense oligonucleotide. Here, we demonstrate the existence of 2 additional poison exons in introns 1 and 22 of SCN1A through targeted, deep-coverage long-read sequencing of SCN1A transcripts. We show that inclusion of these poison exons is developmentally regulated in the human brain, and that deep intronic variants associated with these poison exons lead to their aberrant inclusion in vitro in a minigene assay or in iPSC-derived neurons. Additionally, we show that splice-modulating antisense oligonucleotides can ameliorate aberrant inclusion of poison exons. Our findings highlight the role of deep intronic pathogenic variants in disease and provide additional therapeutic targets for precision medicine in Dravet syndrome and other SCN1A-related disorders.
Keywords: Epilepsy; Gene therapy; Genetics; Neuroscience.
Conflict of interest statement
Figures




References
-
- Scheffer IE, Nabbout R. SCN1A-related phenotypes: epilepsy and beyond. Epilepsia. 2019;60 Suppl 3(s3):S17–S24. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
