

Deconvolution and inference of spatial communication through optimization algorithm for spatial transcriptomics
- PMID: 39948133
- PMCID: PMC11825862
- DOI: 10.1038/s42003-025-07625-8
Deconvolution and inference of spatial communication through optimization algorithm for spatial transcriptomics
Abstract
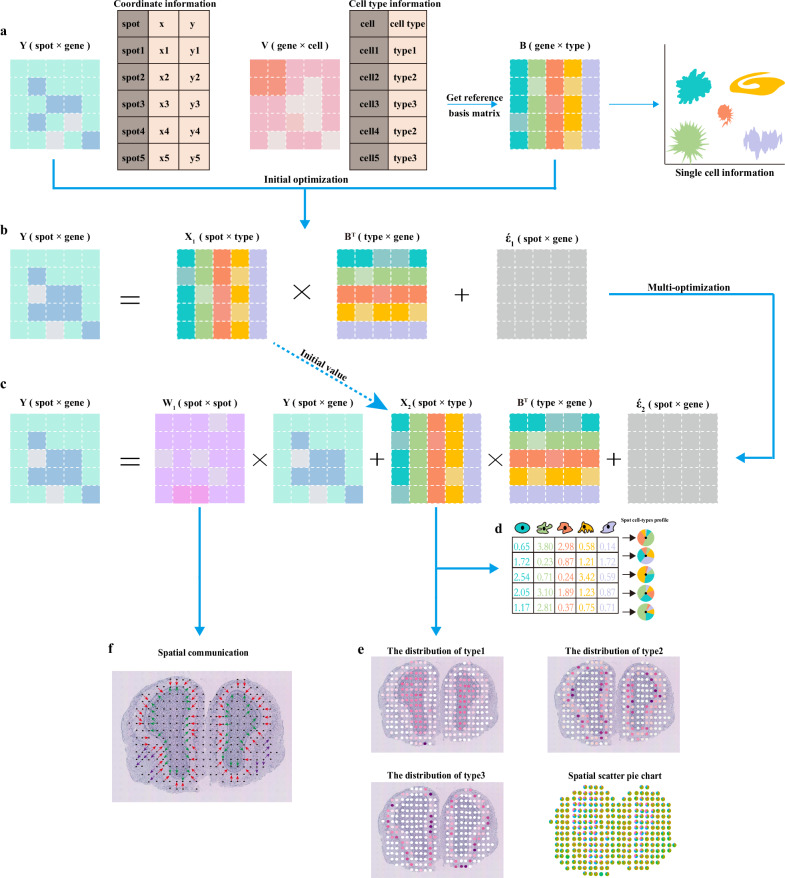
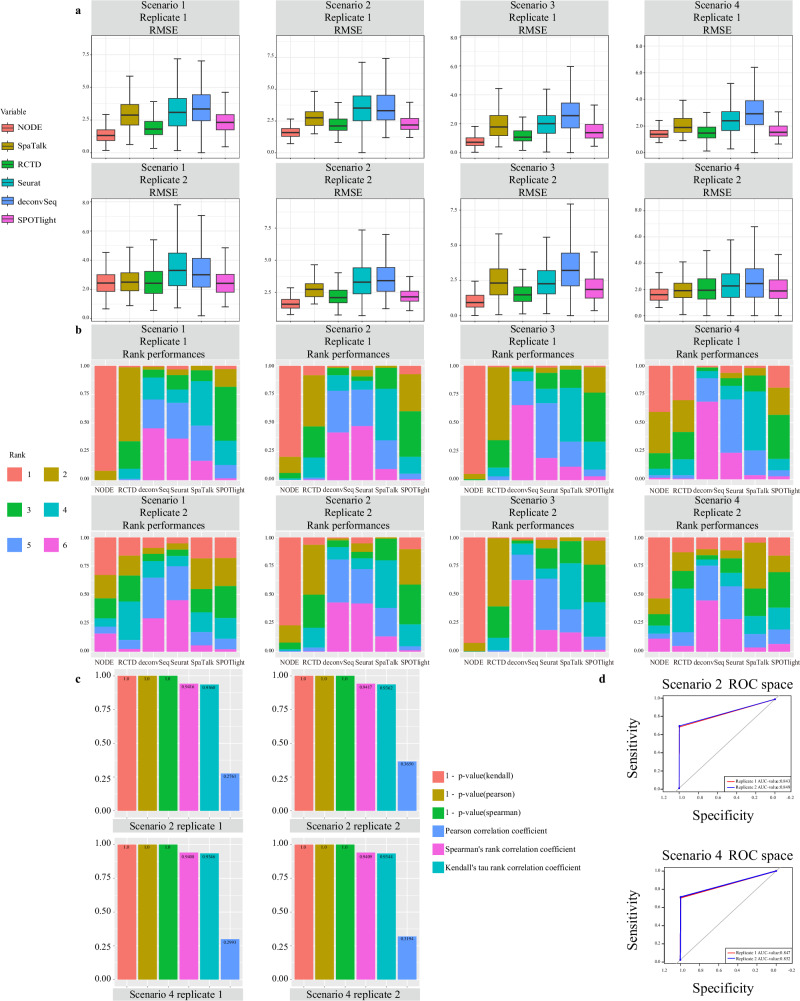
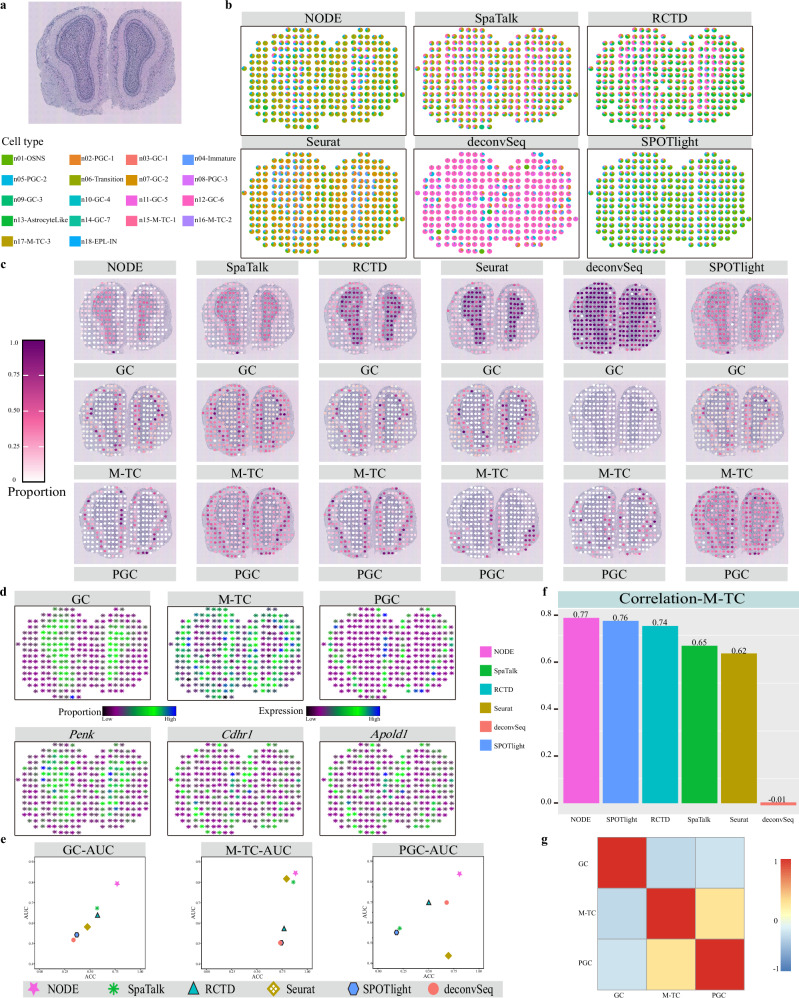
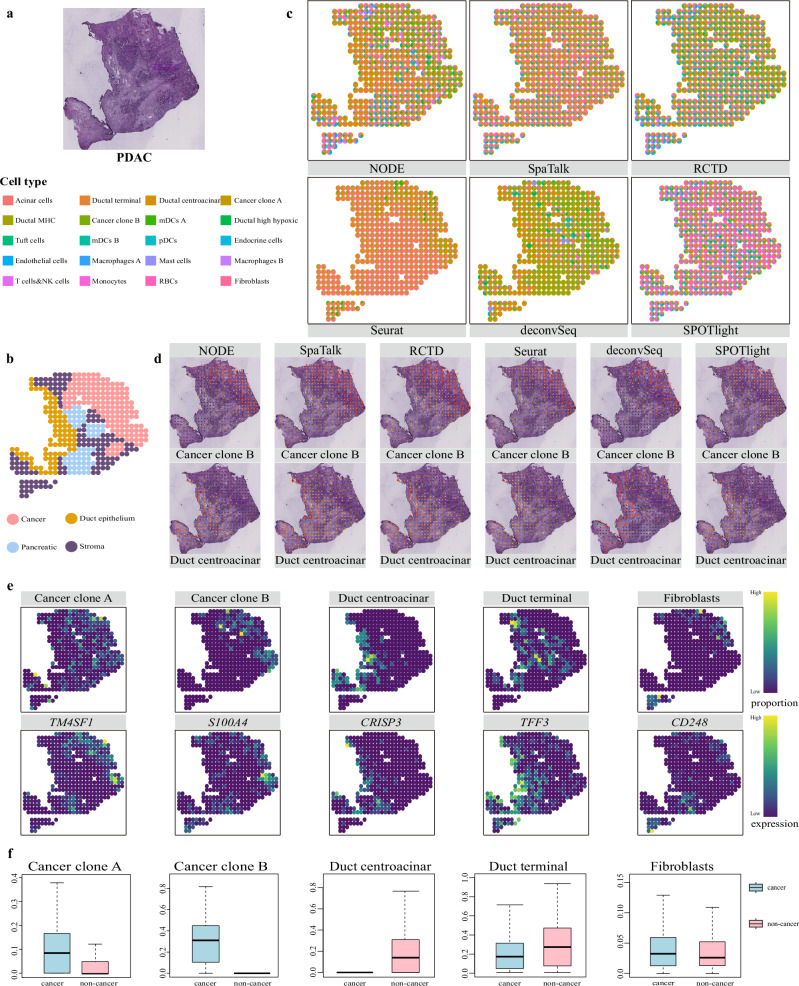
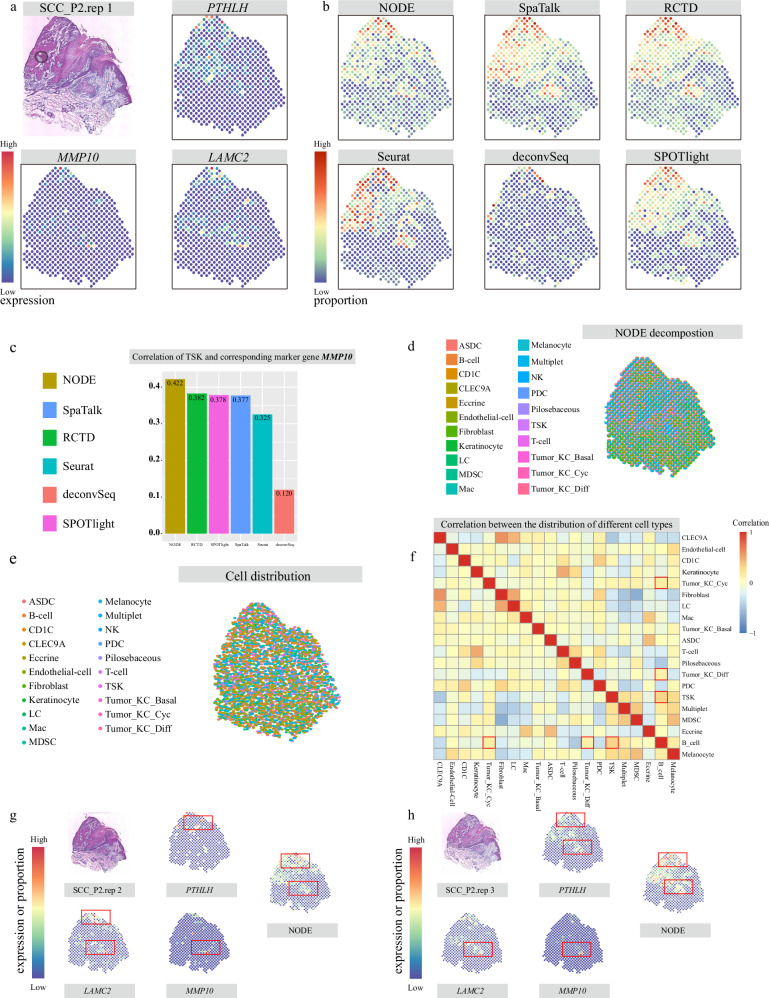
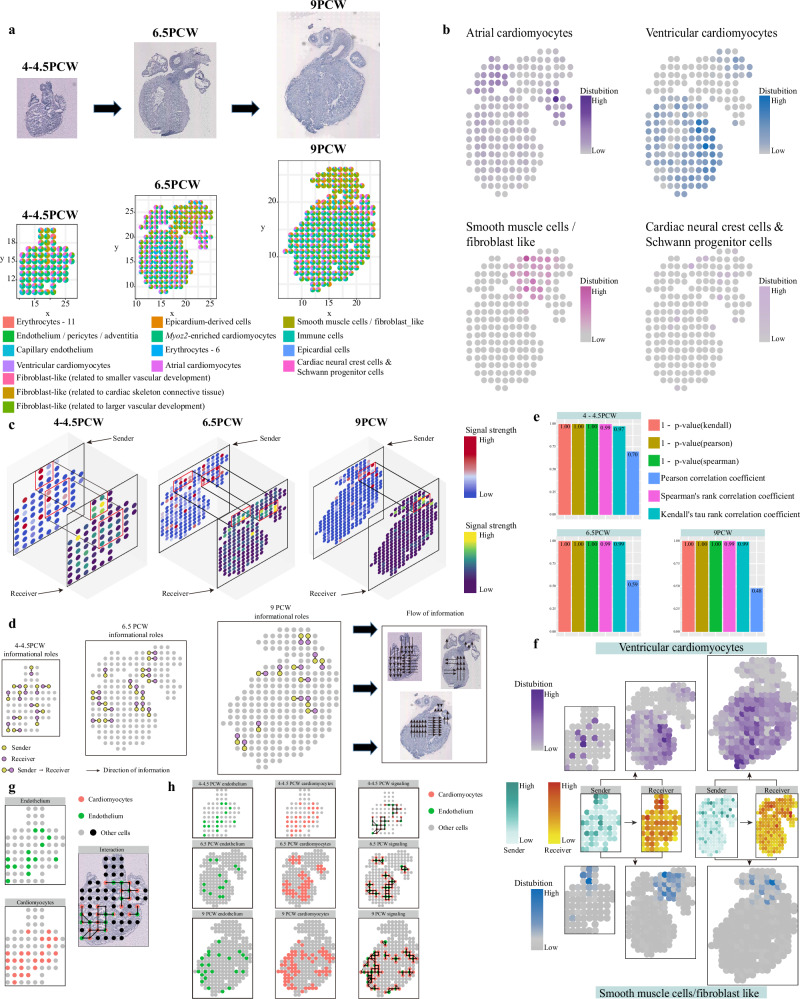
Spatial transcriptomics technologies can capture gene expression at spatial loci. However, at certain resolutions, the obtained gene expression reflects the sum of either a heterogeneous or homogeneous set of cells, rather than individual cell. This limitation gives rise to the deconvolution algorithm to make cell-type inferences at each location. Yet, the vast majority of deconvolution methods that have been developed ignore the spatial information of the tissue and the communications between the cells or spots. To overcome these afflictions, we proposed a deconvolution method, non-negative least squares-based and optimization search-based deconvolution (NODE), that combines cell-type-specific information from single-cell RNA sequencing (scRNA-seq) and intercellular communications in tissue. NODE deconvolution algorithm, incorporating the spatial information of the tissue, allows us to quantify intercellular communications at the same instant. NODE can not only utilize optimization method to infer the deconvolution results of spatial transcriptomics data and reduce the probability of overfitting situations, but also make reasonable inferences for spatial communications. Subsequently, we applied NODE to four datasets to validate the correctness of the NODE deconvolution results and compare them with existing deconvolution algorithms. NODE also inferred spatial communications and validated them in tissue development of human heart.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
Similar articles
-
Precise gene expression deconvolution in spatial transcriptomics with STged.Nucleic Acids Res. 2025 Feb 8;53(4):gkaf087. doi: 10.1093/nar/gkaf087. Nucleic Acids Res. 2025. PMID: 39970279 Free PMC article.
-
SpatialPrompt: spatially aware scalable and accurate tool for spot deconvolution and domain identification in spatial transcriptomics.Commun Biol. 2024 May 25;7(1):639. doi: 10.1038/s42003-024-06349-5. Commun Biol. 2024. PMID: 38796505 Free PMC article.
-
Robust Spatial Cell-Type Deconvolution with Qualitative Reference for Spatial Transcriptomics.Small Methods. 2025 May;9(5):e2401145. doi: 10.1002/smtd.202401145. Epub 2025 Mar 9. Small Methods. 2025. PMID: 40059456 Free PMC article.
-
Computational solutions for spatial transcriptomics.Comput Struct Biotechnol J. 2022 Sep 1;20:4870-4884. doi: 10.1016/j.csbj.2022.08.043. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36147664 Free PMC article. Review.
-
Challenges and opportunities to computationally deconvolve heterogeneous tissue with varying cell sizes using single-cell RNA-sequencing datasets.Genome Biol. 2023 Dec 14;24(1):288. doi: 10.1186/s13059-023-03123-4. Genome Biol. 2023. PMID: 38098055 Free PMC article. Review.
Cited by
-
Epithelial Cell Dysfunction in Pulmonary Fibrosis: Mechanisms, Interactions, and Emerging Therapeutic Targets.Pharmaceuticals (Basel). 2025 May 28;18(6):812. doi: 10.3390/ph18060812. Pharmaceuticals (Basel). 2025. PMID: 40573209 Free PMC article. Review.
References
-
- Burgess, D. J. Spatial transcriptomics coming of age. Nat. Rev. Genet.20, 317–317 (2019). - PubMed
-
- Soldatov, R. et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science (New York, N.Y.)364, 10.1126/science.aas9536 (2019). - PubMed
-
- Prinz, M., Priller, J., Sisodia, S. S. & Ransohoff, R. M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci.14, 1227–1235 (2011). - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
