

Disruption of mitochondrial homeostasis and permeability transition pore opening in OPA1 iPSC-derived retinal ganglion cells
- PMID: 39948685
- PMCID: PMC11823152
- DOI: 10.1186/s40478-025-01942-z
Disruption of mitochondrial homeostasis and permeability transition pore opening in OPA1 iPSC-derived retinal ganglion cells
Abstract
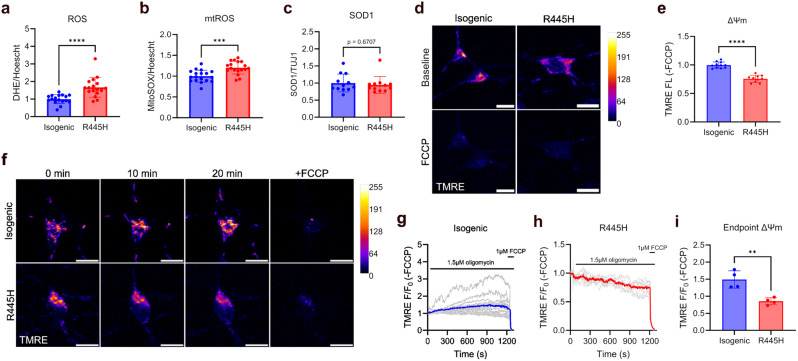
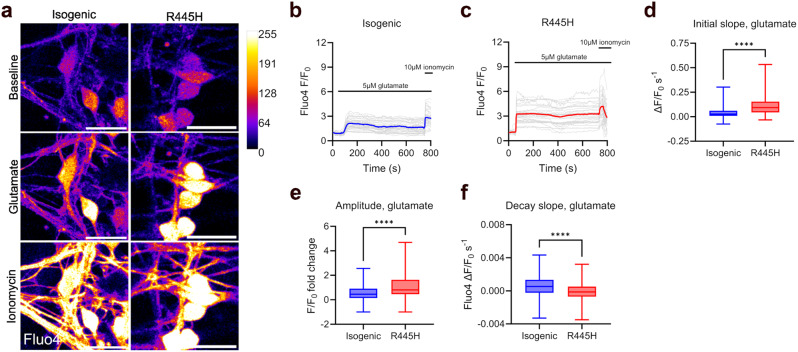
Dominant optic atrophy (DOA) is the most common inherited optic neuropathy, characterised by the selective loss of retinal ganglion cells (RGCs). Over 60% of DOA cases are caused by pathogenic variants in the OPA1 gene, which encodes a dynamin-related GTPase protein. OPA1 plays a key role in the maintenance of the mitochondrial network, mitochondrial DNA integrity and bioenergetic function. However, our current understanding of how OPA1 dysfunction contributes to vision loss in DOA patients has been limited by access to patient-derived RGCs. Here, we used induced pluripotent stem cell (iPSC)-RGCs to study how OPA1 dysfunction affects cellular homeostasis in human RGCs. iPSCs derived from a DOA+ patient with the OPA1 R445H variant and isogenic CRISPR-Cas9-corrected iPSCs were differentiated to iPSC-RGCs. Defects in mitochondrial networks and increased levels of reactive oxygen species were observed in iPSC-RGCs carrying OPA1 R445H. Ultrastructural analyses also revealed changes in mitochondrial shape and cristae structure, with decreased endoplasmic reticulum (ER): mitochondrial contact length in DOA iPSC-RGCs. Mitochondrial membrane potential was reduced and its maintenance was also impaired following inhibition of the F1Fo-ATP synthase with oligomycin, suggesting that mitochondrial membrane potential is maintained in DOA iPSC-RGCs through reversal of the ATP synthase and ATP hydrolysis. These impairments in mitochondrial structure and function were associated with defects in cytosolic calcium buffering following ER calcium release and store-operated calcium entry, and following stimulation with the excitatory neurotransmitter glutamate. In response to mitochondrial calcium overload, DOA iPSC-RGCs exhibited increased sensitivity to opening of the mitochondrial permeability transition pore. These data reveal novel aspects of DOA pathogenesis in R445H patient-derived RGCs. The findings suggest a mechanism in which primary defects in mitochondrial network dynamics disrupt core mitochondrial functions, including bioenergetics, calcium homeostasis, and opening of the permeability transition pore, which may contribute to vision loss in DOA patients.
Keywords: Calcium homeostasis; Dominant optic atrophy; Mitochondrial networks; Neurodegeneration; OPA1; Retinal ganglion cells; iPSCs.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: For the use of patient-derived fibroblasts in this study, informed consent was obtained following the tenets of the Declaration of Helsinki. Ethical approval was granted by the Yorkshire and The Humber-Leeds Bradford Research Ethics Committee (REC reference: 13/YH/0310). Consent for publication: Not applicable. Competing interests: PYWM is a consultant for GenSight Biologics, Stoke Therapeutics, PYC Therapeutics and has received research support from GenSight Biologics and Chiesi. MEC is a consultant for Prime Medicine.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
