

De novo identification of universal cell mechanics gene signatures
- PMID: 39960760
- PMCID: PMC11832173
- DOI: 10.7554/eLife.87930
De novo identification of universal cell mechanics gene signatures
Abstract
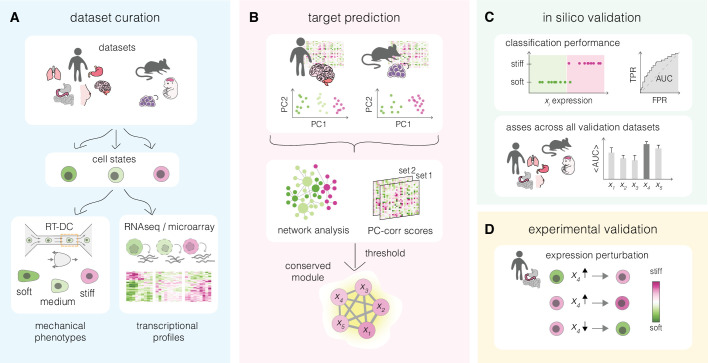
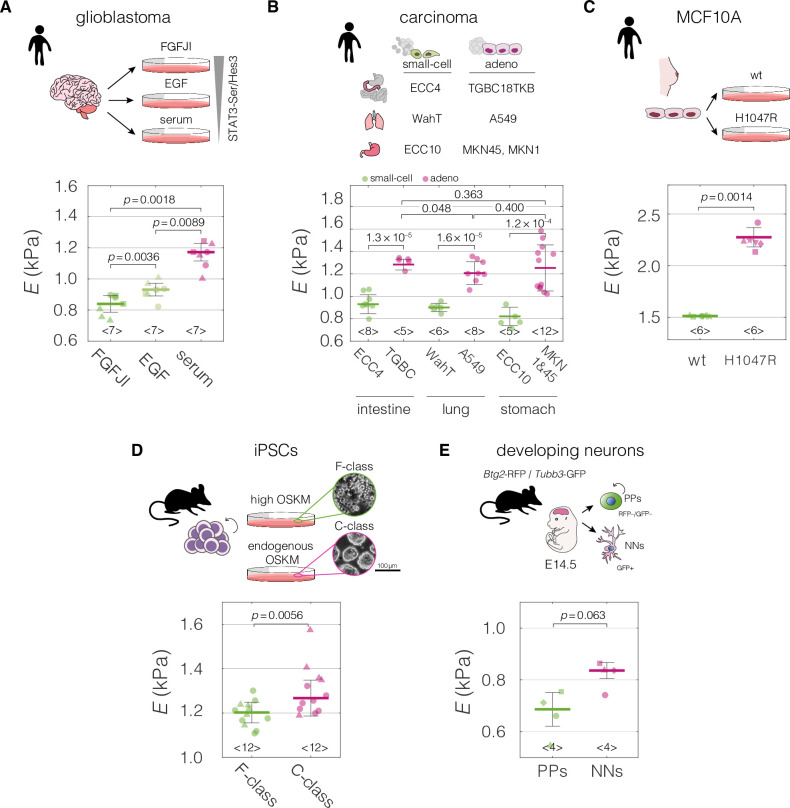
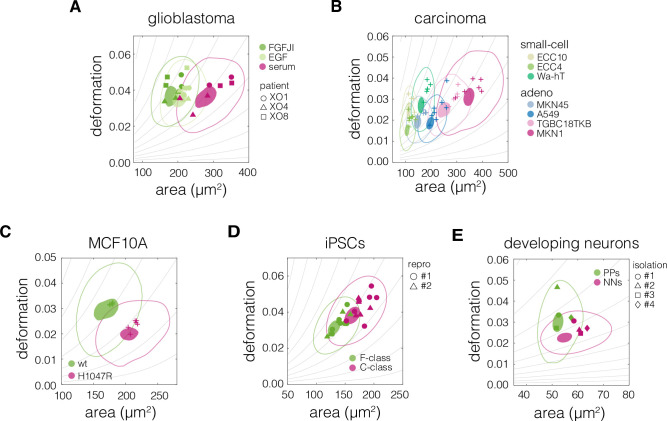
Cell mechanical properties determine many physiological functions, such as cell fate specification, migration, or circulation through vasculature. Identifying factors that govern the mechanical properties is therefore a subject of great interest. Here, we present a mechanomics approach for establishing links between single-cell mechanical phenotype changes and the genes involved in driving them. We combine mechanical characterization of cells across a variety of mouse and human systems with machine learning-based discriminative network analysis of associated transcriptomic profiles to infer a conserved network module of five genes with putative roles in cell mechanics regulation. We validate in silico that the identified gene markers are universal, trustworthy, and specific to the mechanical phenotype across the studied mouse and human systems, and demonstrate experimentally that a selected target, CAV1, changes the mechanical phenotype of cells accordingly when silenced or overexpressed. Our data-driven approach paves the way toward engineering cell mechanical properties on demand to explore their impact on physiological and pathological cell functions.
Keywords: cell biology; deformability cytometry; discriminative network analysis; human; mechanical phenotype; mechanomics; mouse; physics of living systems; transcriptomics; unsupervised machine learning.
© 2023, Urbanska, Ge et al.
Conflict of interest statement
MU, YG, MW, SA, MH, NT, JD, OF, MD, FC, FL, MÁP, AT, CC No competing interests declared, SA, MK, JG Co-founder and shareholder of the company Rivercyte GmbH that is commercializing deformability cytometry technology
Figures



































Update of
- doi: 10.1101/2021.04.26.441418
- doi: 10.7554/eLife.87930.1
- doi: 10.7554/eLife.87930.2
References
-
- Acevedo A, Duran C, Kuo MJ, Ciucci S, Schroeder M, Cannistraci CV. Measuring group separability in geometrical space for evaluation of pattern recognition and dimension reduction algorithms. IEEE Access. 2022;10:22441–22471. doi: 10.1109/ACCESS.2022.3152789. - DOI
-
- Alcaraz J, Buscemi L, Puig-de-Morales M, Colchero J, Baró A, Navajas D. Correction of microrheological measurements of soft samples with atomic force microscopy for the hydrodynamic drag on the cantilever. Langmuir. 2002;18:716–721. doi: 10.1021/la0110850. - DOI
-
- Aprea J, Prenninger S, Dori M, Ghosh T, Monasor LS, Wessendorf E, Zocher S, Massalini S, Alexopoulou D, Lesche M, Dahl A, Groszer M, Hiller M, Calegari F. Transcriptome sequencing during mouse brain development identifies long non-coding RNAs functionally involved in neurogenic commitment. The EMBO Journal. 2013;32:3145–3160. doi: 10.1038/emboj.2013.245. - DOI - PMC - PubMed
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
- 399422891/Deutsche Forschungsgemeinschaft
- Starting Fund/Tsinghua University
- Alexander von Humboldt Professorship/Alexander von Humboldt-Stiftung
- S2018/NMT-4443/Comunidad de Madrid
- 641639/European Commission
- Zhou Yahui Chair Professorship/Tsinghua University
- 201936-30-31/Fundació la Marató de TV3
- WT_/Wellcome Trust/United Kingdom
- ERC Starting Grant "LightTouch" 282060/European Commission
- GU 612/5-1/Deutsche Forschungsgemeinschaft
- 10.35802/224074/WT_/Wellcome Trust/United Kingdom
- 20241710001/Ministry of Science and Technology of the People's Republic of China
LinkOut - more resources
Full Text Sources
