

Type 2 immunity in allergic diseases
- PMID: 39962262
- PMCID: PMC11868591
- DOI: 10.1038/s41423-025-01261-2
Type 2 immunity in allergic diseases
Abstract
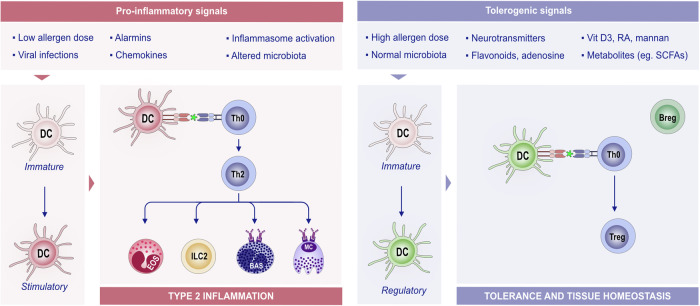
Significant advancements have been made in understanding the cellular and molecular mechanisms of type 2 immunity in allergic diseases such as asthma, allergic rhinitis, chronic rhinosinusitis, eosinophilic esophagitis (EoE), food and drug allergies, and atopic dermatitis (AD). Type 2 immunity has evolved to protect against parasitic diseases and toxins, plays a role in the expulsion of parasites and larvae from inner tissues to the lumen and outside the body, maintains microbe-rich skin and mucosal epithelial barriers and counterbalances the type 1 immune response and its destructive effects. During the development of a type 2 immune response, an innate immune response initiates starting from epithelial cells and innate lymphoid cells (ILCs), including dendritic cells and macrophages, and translates to adaptive T and B-cell immunity, particularly IgE antibody production. Eosinophils, mast cells and basophils have effects on effector functions. Cytokines from ILC2s and CD4+ helper type 2 (Th2) cells, CD8 + T cells, and NK-T cells, along with myeloid cells, including IL-4, IL-5, IL-9, and IL-13, initiate and sustain allergic inflammation via T cell cells, eosinophils, and ILC2s; promote IgE class switching; and open the epithelial barrier. Epithelial cell activation, alarmin release and barrier dysfunction are key in the development of not only allergic diseases but also many other systemic diseases. Recent biologics targeting the pathways and effector functions of IL4/IL13, IL-5, and IgE have shown promising results for almost all ages, although some patients with severe allergic diseases do not respond to these therapies, highlighting the unmet need for a more detailed and personalized approach.
Keywords: Alarmins; allergic diseases; biologics; epithelial barrier; type 2 immunity.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: S.S. is currently a salaried employee of Seed Health, a probiotic retailer. R.D. is a cofounder and CEO in Seed Health. W.V. has received research grants from PROMEDICA Stiftung, Switzerland, and EoE Stiftung, Switzerland, and consulting fees from Mabylon AG, Switzerland. M.S. has received research grants from the Swiss National Science Foundation (SNSF nr 310030_189334/1 and 320030E_224154), GSK, Novartis, Stiftung vorm. Bündner Heilstätte Arosa, Thermofisher and OM Pharma, speaker’s fee from AstraZeneca and consults for Roche. She is a member-at-large of the European Academy of Allergy and Clinical Immunology (EAACI) and EAACI Educational Events Committee Chair. O.P. received research grants from MINECO, Ministerio de Ciencia e Innovación, CAM, Inmunotek S.L., Novartis, and AstraZeneca and fees for giving scientific lectures or participation in Advisory Boards from AstraZeneca, Pfizer, GlaxoSmithKline, Inmunotek S.L., Novartis and Sanofi-Genzyme. K.C.N. reports grants from the National Institute of Allergy and Infectious Diseases (NIAID), National Heart, Lung, and Blood Institute (NHLBI), and from the National Institute of Environmental Health Sciences (NIEHS), other from the Immune Tolerance Network (ITN), other from National Institutes of Health (NIH) clinical research centers, during the conduct of the study; other from IgGenix, other from Seed Health, other from ClostraBio, other from Cour, other from Alladapt, other from Excellergy, other from Red Tree Ventures, other from Regeneron, other from Latitude, outside the submitted work. In addition, K.C.N. has the following patents: “Mixed allergen composition and methods for using the same”, “Granulocyte-based methods for detecting and monitoring immune system disorders”, and “Methods and Assays for Detecting and Quantifying Pure Subpopulations of White Blood Cells in Immune System Disorders”. M.A. has received research grants from the Swiss National Science Foundation, Bern; research grants from Stanford University; Leading House for the Latin American Region; and Seed Money Grants. She is a Scientific Advisory Board member of Stanford University-Sean Parker Asthma Allergy Center, CA; an Advisory Board member of the LEO Foundation Skin Immunology Research Center, Copenhagen; and a Scientific Co-Chair of the World Allergy Congress (WAC) Istanbul, 2022, Scientific Programme Committee Chair, EAACI. C.A.A. has received research grants from the Swiss National Science Foundation, European Union (EU CURE, EU Syn-Air-G), Novartis Research Institutes (Basel, Switzerland), Stanford University (Redwood City, Calif), Seed Health (Boston, USA), AO Research Institute (Davos, Switzerland) and SciBase (Stockholm, Sweden). He is the cochair for the EAACI Guidelines on Environmental Science in Allergic Diseases and Asthma; Chair of the EAACI Epithelial Cell Biology Working Group. Serves on the Advisory Boards of Sanofi/Regeneron (Bern, Switzerland, New York, USA), Stanford University Sean Parker Asthma Allergy Center (CA, USA), Novartis (Basel, Switzerland), GlaxoSmithKline (Zurich, Switzerland), Bristol-Myers Squibb (New York, USA), Seed Health (Boston, USA) and SciBase (Stockholm, Sweden). C.A.A. is the Editor-in-Chief of Allergy. I.O., Y.M., D.Y., Y.P., S.A., M.L., P.D’A., C.B., H.B., B.Z., C.Z., O.G.V., O.A., A.K., A.G.-S., J.-F.L., L.S., M.Y., S.R.S., U.R., A.J.K., M.B.I, and M.M.-F. declare no relevant conflicts of interest.
Figures









References
-
- Akdis CA. Does the epithelial barrier hypothesis explain the increase in allergy, autoimmunity and other chronic conditions? Nat Rev Immunol. 2021;21:739–51. - PubMed
-
- Kolkhir P, Akdis CA, Akdis M, Bachert C, Bieber T, Canonica GW, et al. Type 2 chronic inflammatory diseases: targets, therapies and unmet needs. Nat Rev Drug Discov. 2023;22:743–67. - PubMed
-
- Akdis CA, Arkwright PD, Bruggen MC, Busse W, Gadina M, Guttman-Yassky E, et al. Type 2 immunity in the skin and lungs. Allergy. 2020;75:1582–605. - PubMed
-
- Trautmann A, Schmid-Grendelmeier P, Kruger K, Crameri R, Akdis M, Akkaya A, et al. T cells and eosinophils cooperate in the induction of bronchial epithelial cell apoptosis in asthma. J Allergy Clin Immunol. 2002;109:329–37. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
