Super-enhancer-driven EFNA1 fuels tumor progression in cervical cancer via the FOSL2-Src/AKT/STAT3 axis
- PMID: 39964764
- PMCID: PMC11996870
- DOI: 10.1172/JCI177599
Super-enhancer-driven EFNA1 fuels tumor progression in cervical cancer via the FOSL2-Src/AKT/STAT3 axis
Abstract
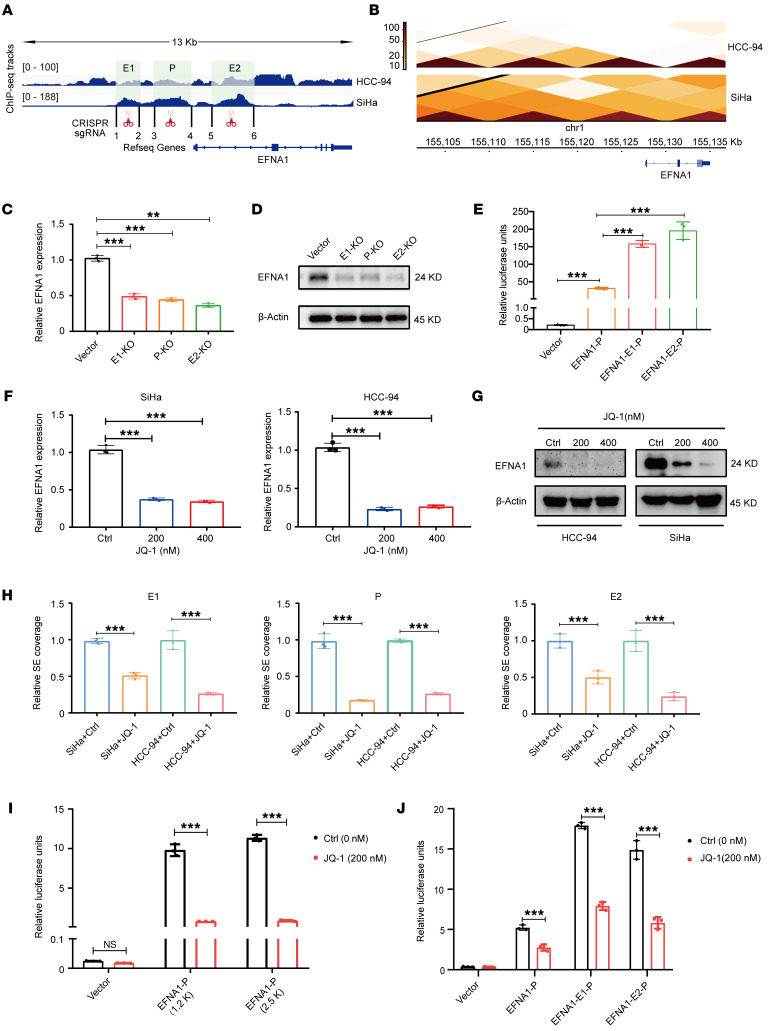
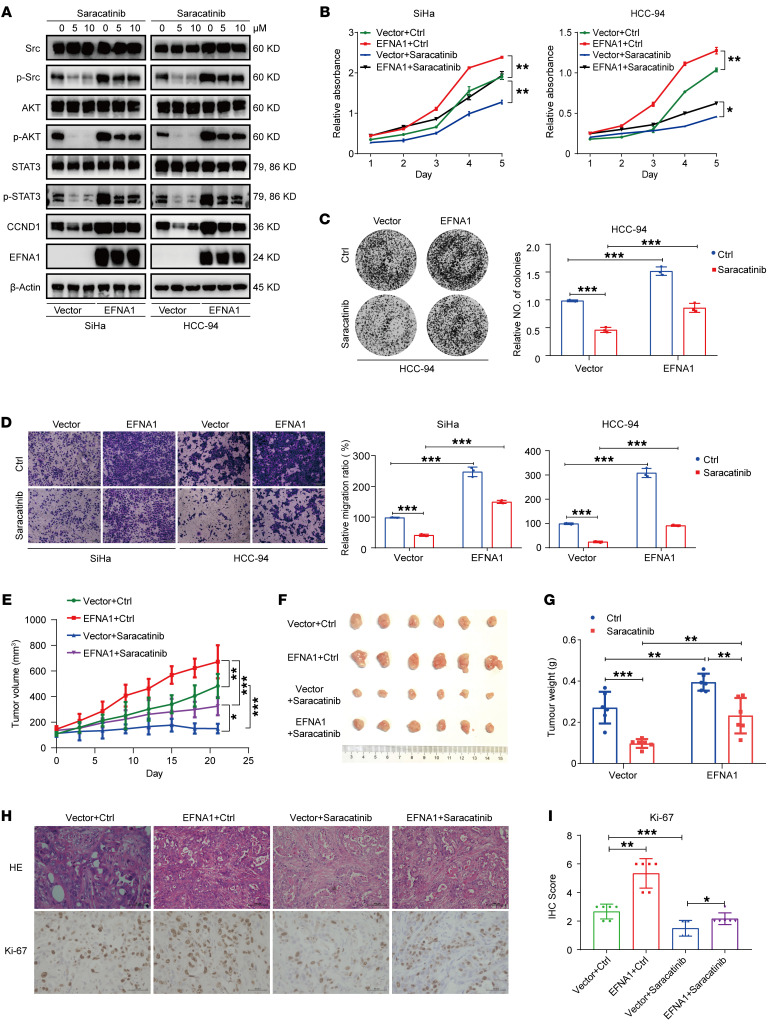
Super-enhancers (SEs) are expansive cis-regulatory elements known for amplifying oncogene expression across various cancers. However, their role in cervical cancer (CC), a remarkable global malignancy affecting women, remains underexplored. Here we applied integrated epigenomic and transcriptomic profiling to delineate the distinct SE landscape in CC by analyzing paired tumor and normal tissues. Our study identifies a tumor-specific SE at the EFNA1 locus that drives EFNA1 expression in CC. Mechanically, the EFNA1-SE region contains consensus sequences for the transcription factor FOSL2, whose knockdown markedly suppressed luciferase activity and diminished H3K27ac enrichment within the SE region. Functional analyses further underlined EFNA1's oncogenic role in CC, linking its overexpression to poor patient outcomes. EFNA1 knockdown strikingly reduced CC cell proliferation, migration, and tumor growth. Moreover, EFNA1 cis-interacted with its receptor EphA2, leading to decreased EphA2 tyrosine phosphorylation and subsequent activation of the Src/AKT/STAT3 forward signaling pathway. Inhibition of this pathway with specific inhibitors substantially attenuated the tumorigenic capacity of EFNA1-overexpressing CC cells in both in vitro and in vivo models. Collectively, our study unveils the critical role of SEs in promoting tumor progression through the FOSL2-EFNA1-EphA2-Src/AKT/STAT3 axis, providing new prognostic and therapeutic avenues for CC patients.
Keywords: Cell biology; Cervical cancer; Epigenetics; Oncology.
Figures