

Long-read single-cell RNA sequencing enables the study of cancer subclone-specific genotypes and phenotypes in chronic lymphocytic leukemia
- PMID: 39965935
- PMCID: PMC12047255
- DOI: 10.1101/gr.279049.124
Long-read single-cell RNA sequencing enables the study of cancer subclone-specific genotypes and phenotypes in chronic lymphocytic leukemia
Abstract
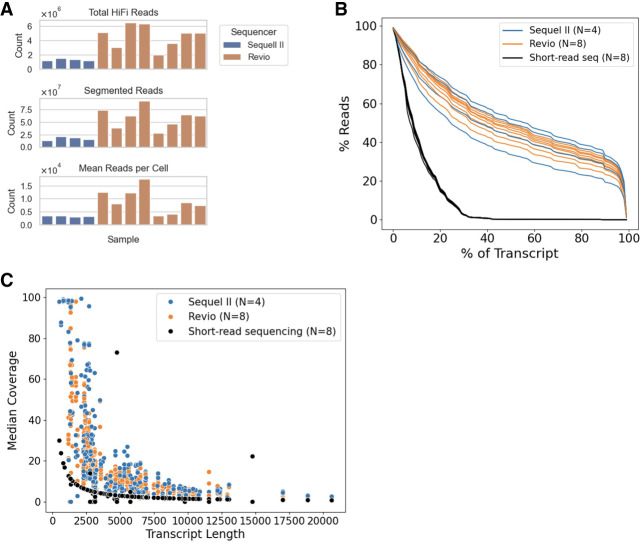
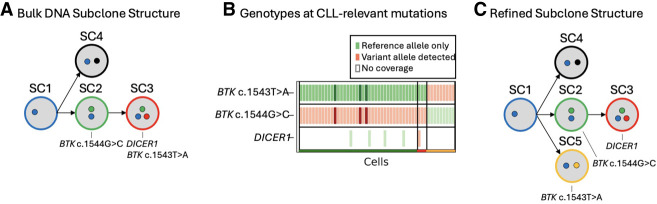
Bruton tyrosine kinase (BTK) inhibitors are effective for the treatment of chronic lymphocytic leukemia (CLL) due to BTK's role in B cell survival and proliferation. Treatment resistance is most commonly caused by the emergence of the hallmark BTK C481S mutation that inhibits drug binding. In this study, we aimed to investigate cancer subclones harboring a BTK C481S mutation and identify cells with co-occurring CLL driver mutations. In addition, we sought to determine whether BTK-mutated subclones exhibit distinct transcriptomic behavior when compared to other cancer subclones. To achieve these goals, we use scBayes, which integrates bulk DNA sequencing and single-cell RNA sequencing (scRNA-seq) data to genotype individual cells for subclone-defining mutations. Although the most common approach for scRNA-seq includes short-read sequencing, transcript coverage is limited due to the vast majority of the reads being concentrated at the priming end of the transcript. Here, we utilized MAS-seq, a long-read scRNA-seq technology, to substantially increase transcript coverage and expand the set of informative mutations to link cells to cancer subclones in six CLL patients who acquired BTK C481S mutations during BTK inhibitor treatment. In two patients who developed two independent BTK-mutated subclones, we find that most BTK-mutated cells have an additional CLL driver gene mutation. When examining subclone-specific gene expression, we find that in one patient, BTK-mutated subclones are transcriptionally distinct from the rest of the malignant B cell population with an overexpression of CLL-relevant genes.
© 2025 Black et al.; Published by Cold Spring Harbor Laboratory Press.
Figures





Update of
-
Long-read single-cell RNA sequencing enables the study of cancer subclone-specific genotype and phenotype in chronic lymphocytic leukemia.bioRxiv [Preprint]. 2024 Mar 16:2024.03.15.585298. doi: 10.1101/2024.03.15.585298. bioRxiv. 2024. Update in: Genome Res. 2025 Apr 14;35(4):686-697. doi: 10.1101/gr.279049.124. PMID: 38559060 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources