

Statistical framework for calling allelic imbalance in high-throughput sequencing data
- PMID: 39966391
- PMCID: PMC11836314
- DOI: 10.1038/s41467-024-55513-2
Statistical framework for calling allelic imbalance in high-throughput sequencing data
Abstract
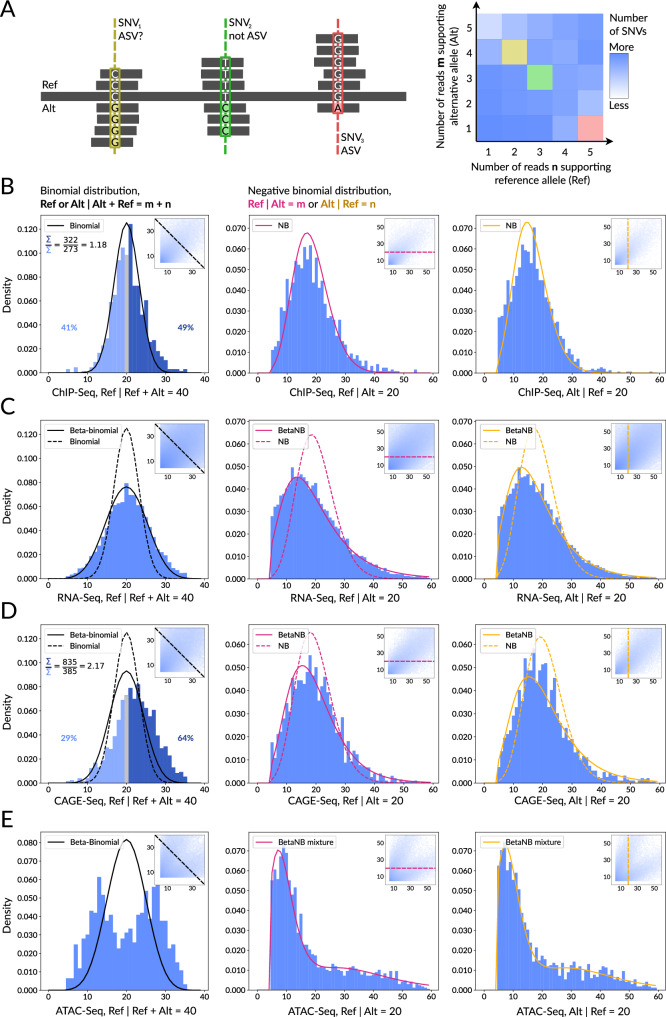
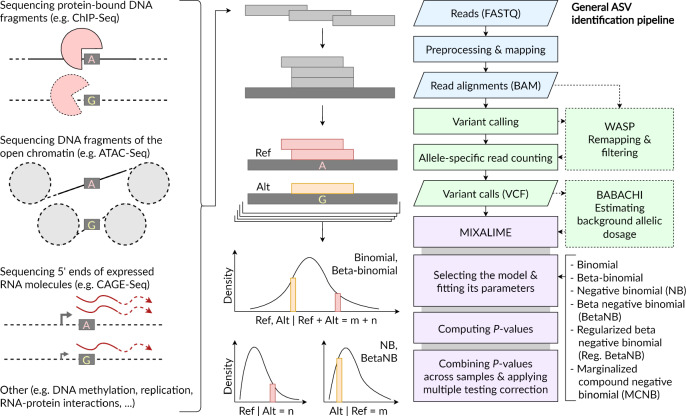
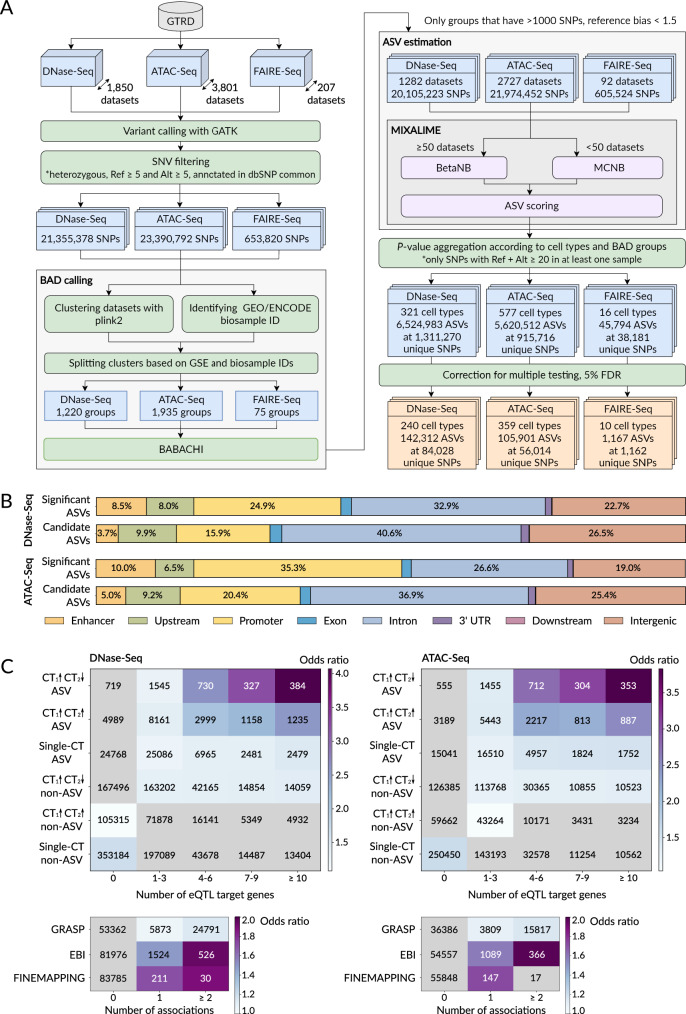
High-throughput sequencing facilitates large-scale studies of gene regulation and allows tracing the associations of individual genomic variants with changes in gene regulation and expression. Compared to classic association studies, the assessment of an allelic imbalance at heterozygous variants captures functional variant effects with smaller sample sizes, higher sensitivity, and better resolution. Yet, identification of allele-specific variants from allelic read counts remains challenging due to data-dependent biases and overdispersion arising from technical and biological variability. We present MIXALIME, a novel computational framework for calling allele-specific variants in diverse omics data with a repertoire of statistical models accounting for read mapping bias and copy number variation. We benchmark MIXALIME with DNase-Seq, ATAC-Seq, and CAGE-Seq data, and we demonstrate that the allelic imbalance highlights causal variants in GWAS results. Finally, as a showcase of the large-scale practical application of MIXALIME, we present an atlas of variants exhibiting allele-specific chromatin accessibility, built from thousands of available datasets obtained from diverse cell types.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures



References
-
- Uffelmann, E. et al. Genome-wide association studies. Nat. Rev. Methods Prim.1, 1–21 (2021).
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
