

Mass spectrometry methods and mathematical PK/PD model for decision tree-guided covalent drug development
- PMID: 39971904
- PMCID: PMC11839910
- DOI: 10.1038/s41467-025-56985-6
Mass spectrometry methods and mathematical PK/PD model for decision tree-guided covalent drug development
Abstract
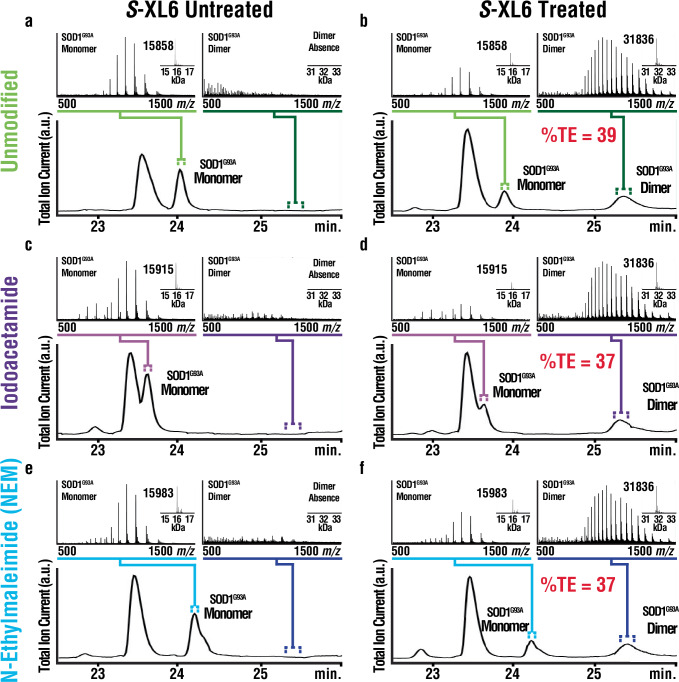
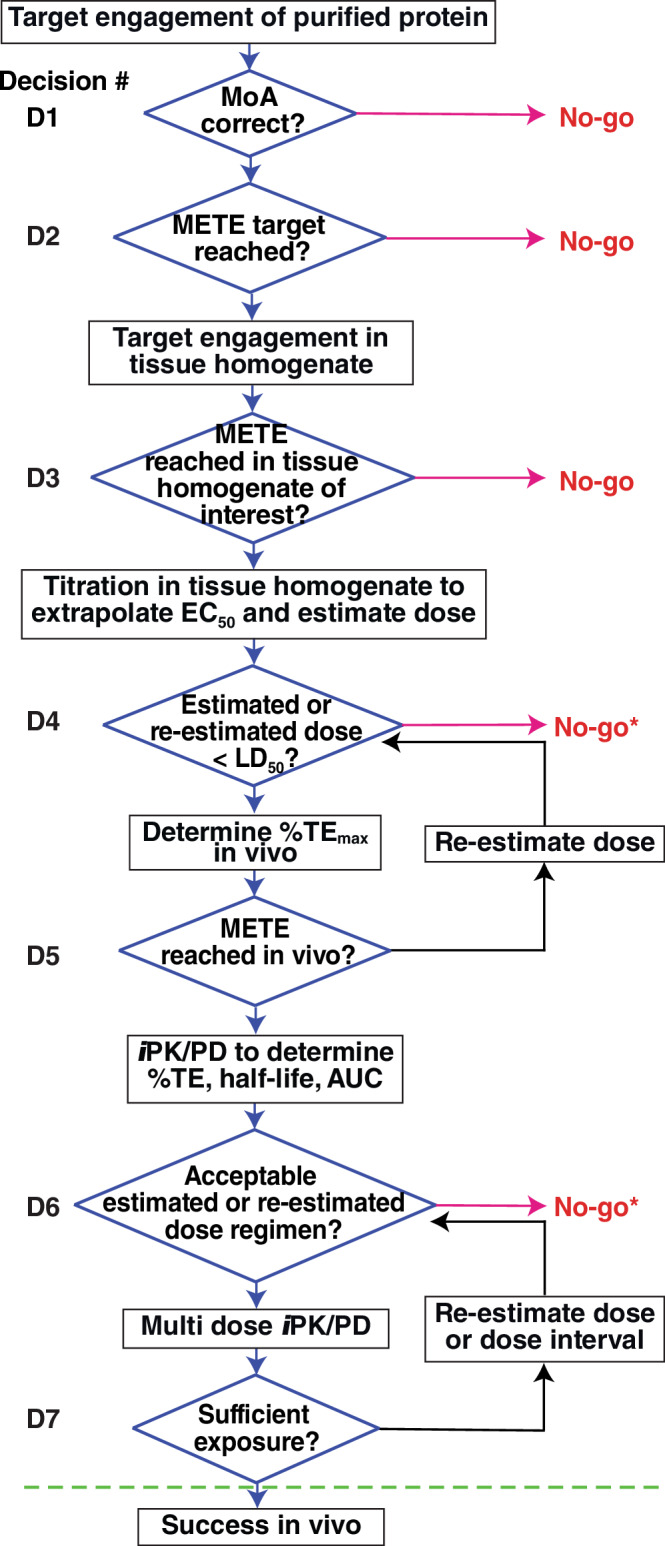
Covalent drug discovery efforts are growing rapidly but have major unaddressed limitations. These include high false positive rates during hit-to-lead identification; the inherent uncoupling of covalent drug concentration and effect [i.e., uncoupling of pharmacokinetics (PK) and pharmacodynamics (PD)]; and a lack of bioanalytical and modeling methods for determining PK and PD parameters. We present a covalent drug discovery workflow that addresses these limitations. Our bioanalytical methods are based upon a mass spectrometry (MS) assay that can measure the percentage of drug-target protein conjugation (% target engagement) in biological matrices. Further we develop an intact protein PK/PD model (iPK/PD) that outputs PK parameters (absorption and distribution) as well as PD parameters (mechanism of action, protein metabolic half-lives, dose, regimen, effect) based on time-dependent target engagement data. Notably, the iPK/PD model is applicable to any measurement (e.g., bottom-up MS and other drug binding studies) that yields % of target engaged. A Decision Tree is presented to guide researchers through the covalent drug development process. Our bioanalytical methods and the Decision Tree are applied to two approved drugs (ibrutinib and sotorasib); the most common plasma off-target, human serum albumin; three protein targets (KRAS, BTK, SOD1), and to a promising SOD1-targeting ALS drug candidates.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: Authors declare no competing interests.
Figures




References
-
- Singh, J. The ascension of targeted covalent inhibitors. J. Med. Chem.65, 5886–5901 (2022). - PubMed
-
- Singh, J., Petter, R. C., Baillie, T. A. & Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov.10, 307–317 (2011). - PubMed
-
- Pichler, C. M., Krysiak, J. & Breinbauer, R. Target identification of covalently binding drugs by activity-based protein profiling (ABPP). Bioorg. Med. Chem.24, 3291–3303 (2016). - PubMed
-
- Deng, H., Lei, Q., Wu, Y., He, Y. & Li, W. Activity-based protein profiling: recent advances in medicinal chemistry. Eur. J. Med. Chem.191, 112151 (2020). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
