

GTPBP2 in-frame deletion in canine model with non-syndromic progressive retinal atrophy
- PMID: 39971978
- PMCID: PMC11840026
- DOI: 10.1038/s41598-025-89446-7
GTPBP2 in-frame deletion in canine model with non-syndromic progressive retinal atrophy
Abstract
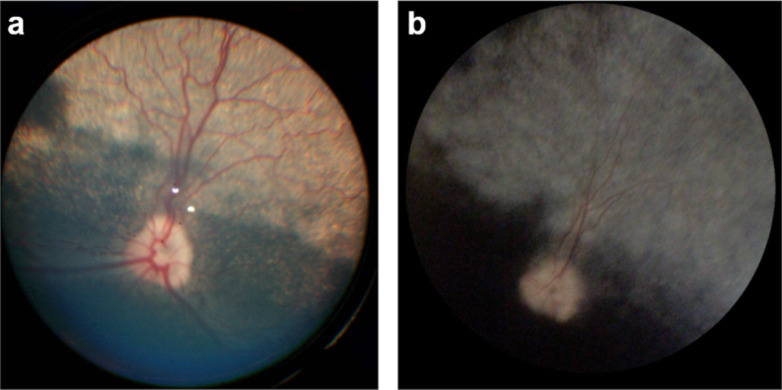
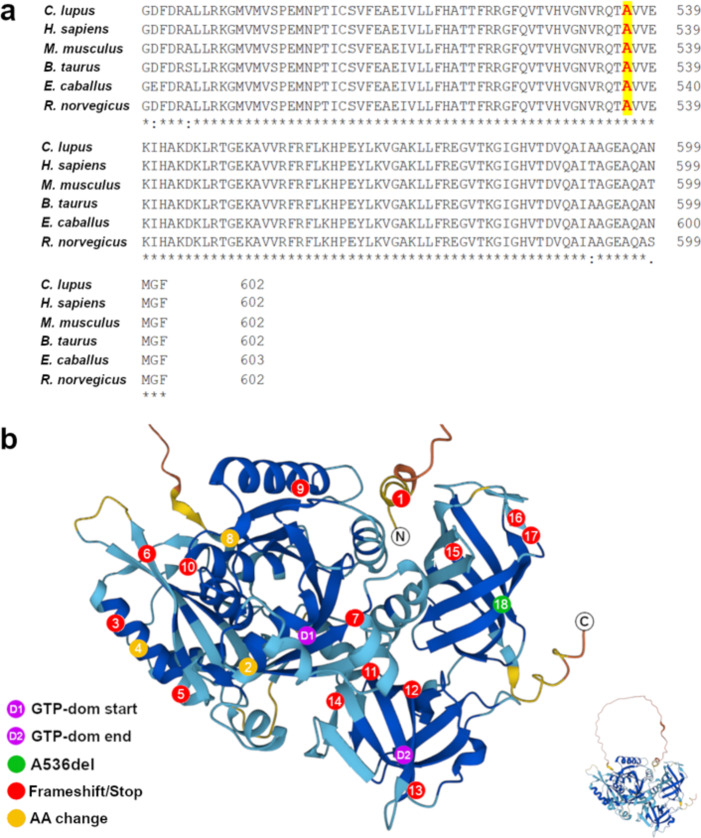
Progressive retinal atrophy (PRA), caused by aberrant functioning of rod/cone photoreceptors, leads to blindness affecting mammals, including dogs. We identified a litter of three Labrador retrievers affected by non-syndromic PRA; the parents and three other siblings were unaffected. Homozygosity mapping and whole-genome sequencing detected a homozygous 3-bp deletion in the coding region of GTPBP2, located in CFA12 (NC_049233.1:12,264,348_12,264,350del, c.1606_1608del, p.Ala536del). The variant was absent from the online European Variation Archive (EVA) database, the Dog Biomedical Variants Database Consortium, and the Dog10k database. We tested 91 non-affected dogs from the same kennel and found 75 wild-type (WT) and 16 carriers, all clinically normal, and 569 Labradors from the general population (USA), all WT. GTPBP2 is associated with Jaberi-Elahi syndrome (JES) in Homo sapiens, and splice variants in Mus musculus are associated with neurodegeneration; in both cases photoreceptor degeneration may be included in its manifestation. Heterologous cellular systems were transfected with cDNA encoding WT or A536del mutant GTPBP2 protein and immunoblot analysis of total cell lysate with anti-GTPBP2 antibodies showed that the expression level of the GTPBP2 mutant protein A536del is slightly but not significantly reduced compared to WT. Immunofluorescent methods and confocal analysis of cells transfected with WT or A536del GTPBP2 protein revealed that the WT form is diffuse throughout the cytosol, while the mutant form resulted in the formation of cytoplasmic aggregates in ~70-80% of cells. The deleted amino acid falls within a conserved interval outside the GTP domain of GTPBP2, suggesting a potentially novel role of the sequence on cellular localization of the protein.
Keywords: Animal model; Non-syndromic; Phenotype variability; Progressive retinal atrophy; Protein domain; Protein function.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: The authors declare no competing interests. Ethical statement: The research and the eye exams were conducted in full compliance and strict accordance with the Association for Research in Vision and Ophthalmology (ARVO) “Resolution on the Use of Animals in Ophthalmic and Vision Research”, and the study protocol was approved by the “Institutional Animal Care and Use Committee” (IACUCs), University of Pennsylvania (code 806301), and were performed in accordance with relevant guidelines and regulations. Eye examinations were carried out by ACVO board certificated veterinary ophthalmologists (GDA, and diplomates who referred the cases). All methods are reported in accordance with ARRIVE guidelines.
Figures




Similar articles
-
Complex Structural PPT1 Variant Associated with Non-syndromic Canine Retinal Degeneration.G3 (Bethesda). 2019 Feb 7;9(2):425-437. doi: 10.1534/g3.118.200859. G3 (Bethesda). 2019. PMID: 30541930 Free PMC article.
-
CCDC66 frameshift variant associated with a new form of early-onset progressive retinal atrophy in Portuguese Water Dogs.Sci Rep. 2020 Dec 3;10(1):21162. doi: 10.1038/s41598-020-77980-5. Sci Rep. 2020. PMID: 33273526 Free PMC article.
-
A LINE-1 insertion situated in the promoter of IMPG2 is associated with autosomal recessive progressive retinal atrophy in Lhasa Apso dogs.BMC Genet. 2020 Sep 7;21(1):100. doi: 10.1186/s12863-020-00911-w. BMC Genet. 2020. PMID: 32894063 Free PMC article.
-
Jaberi-Elahi syndrome: Exploring a novel GTPBP2 mutation and a literature review.Eur J Med Genet. 2024 Aug;70:104953. doi: 10.1016/j.ejmg.2024.104953. Epub 2024 Jun 7. Eur J Med Genet. 2024. PMID: 38852771 Review.
-
A review of research to elucidate the causes of the generalized progressive retinal atrophies.Vet J. 1998 Jan;155(1):5-18. doi: 10.1016/s1090-0233(98)80028-2. Vet J. 1998. PMID: 9455155 Review.
References
-
- Campochiaro, P. A. & Mir, T. A. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res.62, 24–37. 10.1016/j.preteyeres.2017.08.004 (2018). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
