

Human-correlated genetic models identify precision therapy for liver cancer
- PMID: 39972137
- PMCID: PMC11922762
- DOI: 10.1038/s41586-025-08585-z
Human-correlated genetic models identify precision therapy for liver cancer
Abstract
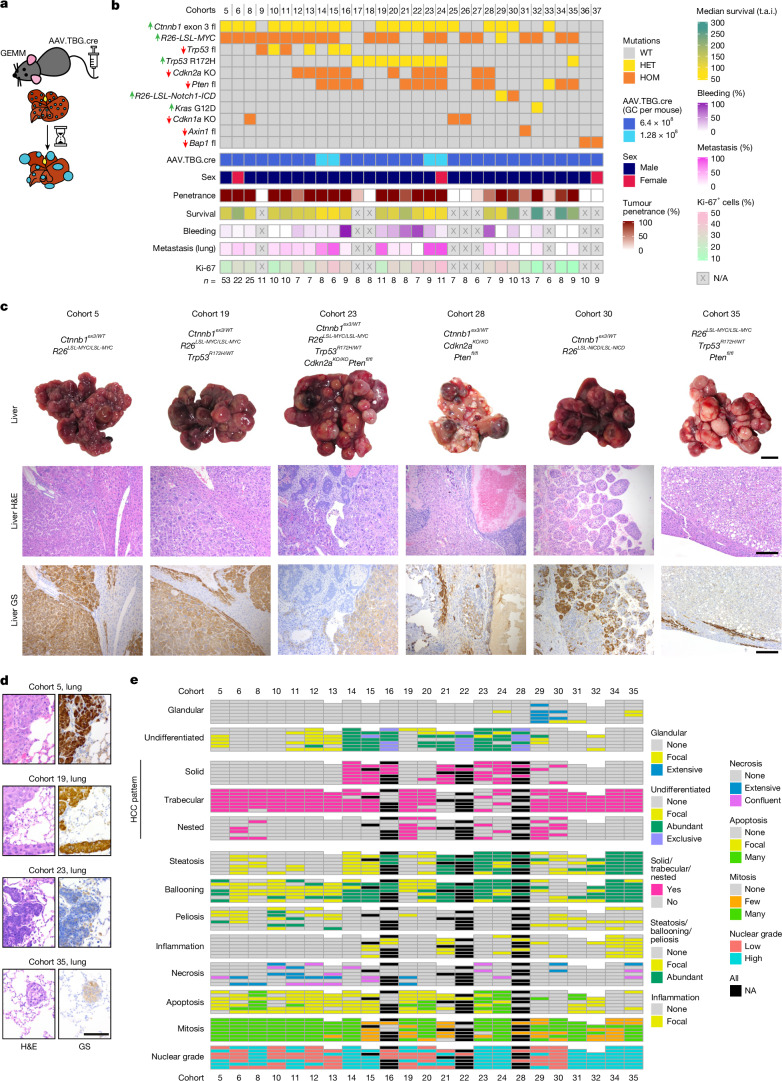
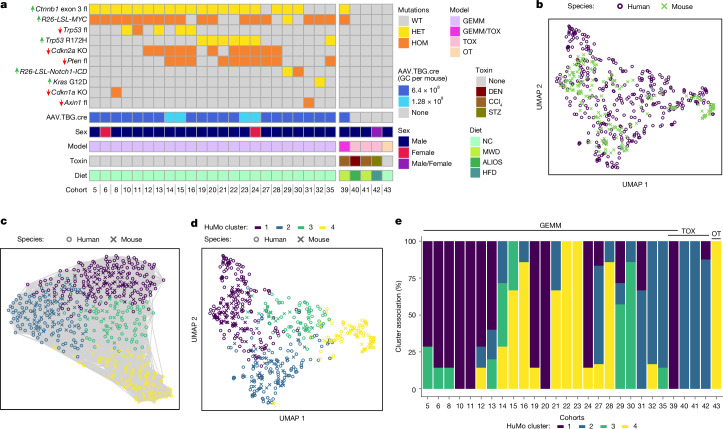
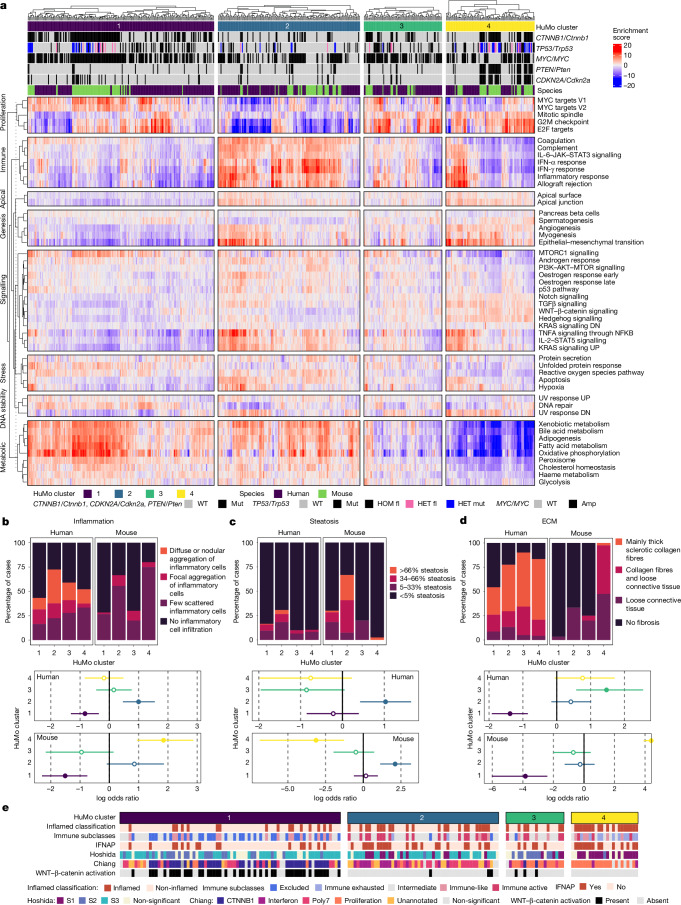
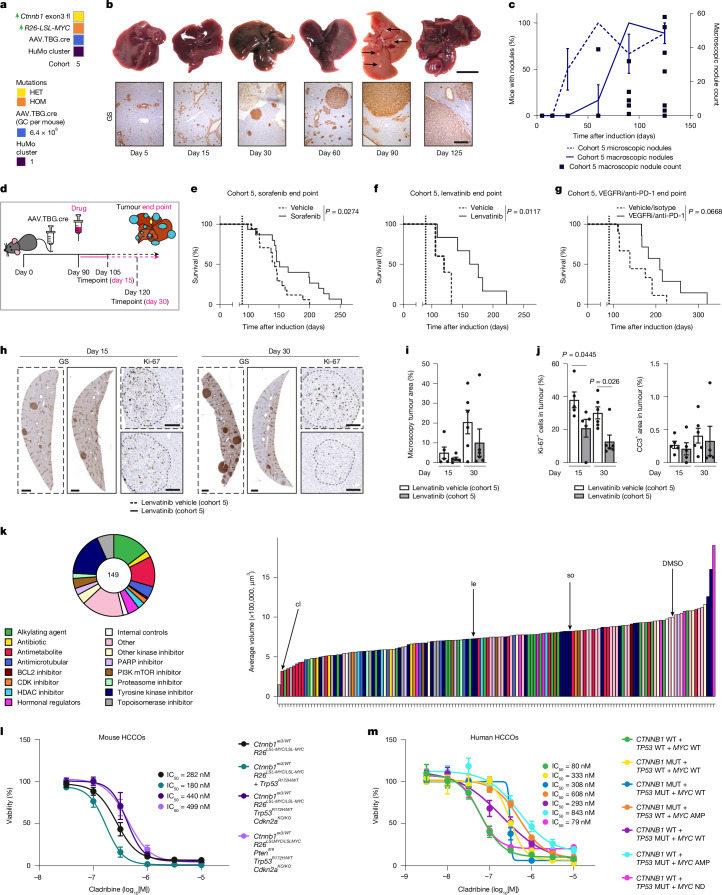
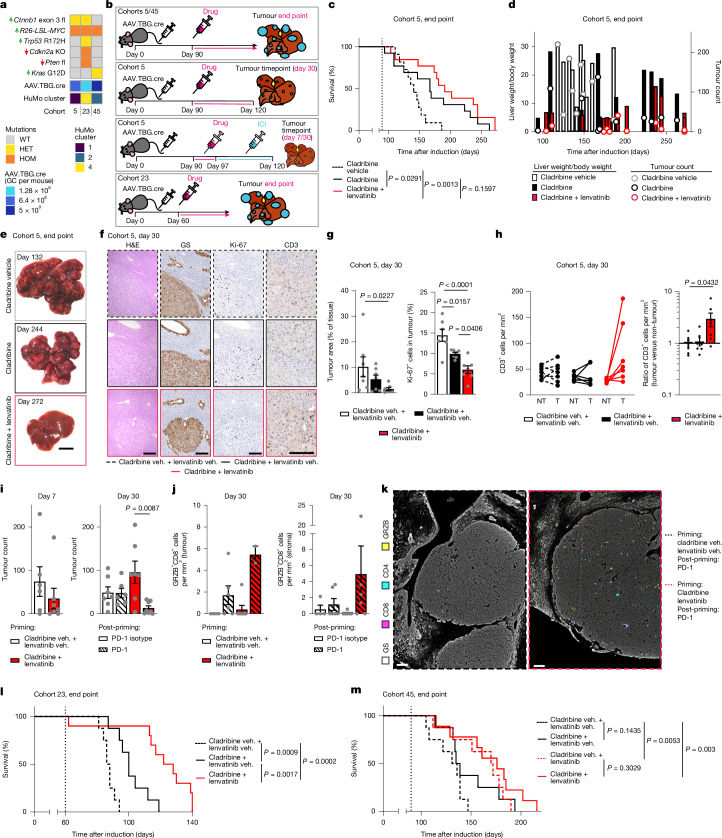
Hepatocellular carcinoma (HCC), the most common form of primary liver cancer, is a leading cause of cancer-related mortality worldwide1,2. HCC occurs typically from a background of chronic liver disease, caused by a spectrum of predisposing conditions. Tumour development is driven by the expansion of clones that accumulate progressive driver mutations3, with hepatocytes the most likely cell of origin2. However, the landscape of driver mutations in HCC is broadly independent of the underlying aetiologies4. Despite an increasing range of systemic treatment options for advanced HCC, outcomes remain heterogeneous and typically poor. Emerging data suggest that drug efficacies depend on disease aetiology and genetic alterations5,6. Exploring subtypes in preclinical models with human relevance will therefore be essential to advance precision medicine in HCC7. Here we generated a suite of genetically driven immunocompetent in vivo and matched in vitro HCC models. Our models represent multiple features of human HCC, including clonal origin, histopathological appearance and metastasis. We integrated transcriptomic data from the mouse models with human HCC data and identified four common human-mouse subtype clusters. The subtype clusters had distinct transcriptomic characteristics that aligned with the human histopathology. In a proof-of-principle analysis, we verified response to standard-of-care treatment and used a linked in vitro-in vivo pipeline to identify a promising therapeutic candidate, cladribine, that has not previously been linked to HCC treatment. Cladribine acts in a highly effective subtype-specific manner in combination with standard-of-care therapy.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: Material for the lenvatinib day 15 and day 30 timepoint experiments (Fig. 4h–j) was provided by Eisai. AZD2171 was provided by AstraZeneca. D.A.M. is a director, shareholder and employee of FibroFind. L.M.C. has consulted for Ono Pharmaceuticals UK on unrelated work. J.M.L. is receiving research support from Eisai Inc and Bayer Pharmaceuticals; is consulting and performing sponsored lectures for Eisai Inc., Merck, Roche, Genentech, AstraZeneca, Bayer Pharmaceuticals, AbbVie, Sanofi, Moderna, Glycotest and Exelixis; and participates in the Data Safety Monitoring Board for Bristol Myers Squibb. The other authors declare no competing interests.
Figures












References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
