

Tumour-wide RNA splicing aberrations generate actionable public neoantigens
- PMID: 39972144
- PMCID: PMC11903331
- DOI: 10.1038/s41586-024-08552-0
Tumour-wide RNA splicing aberrations generate actionable public neoantigens
Abstract
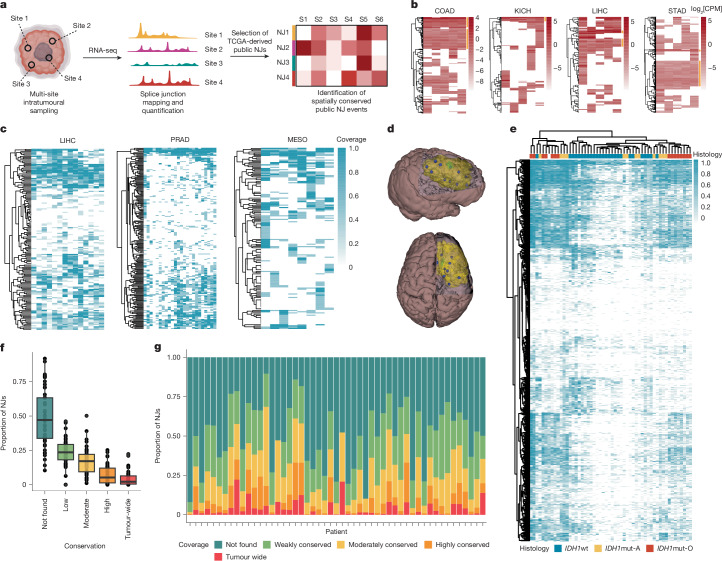
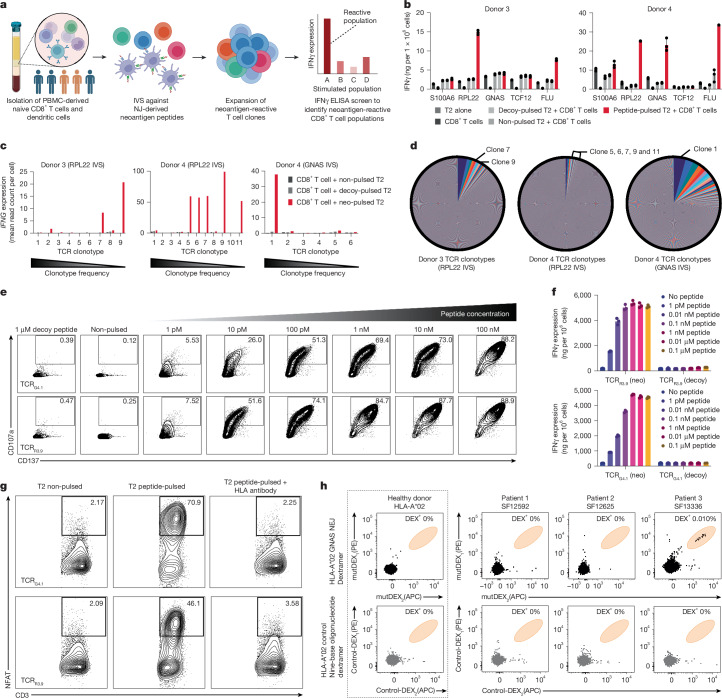
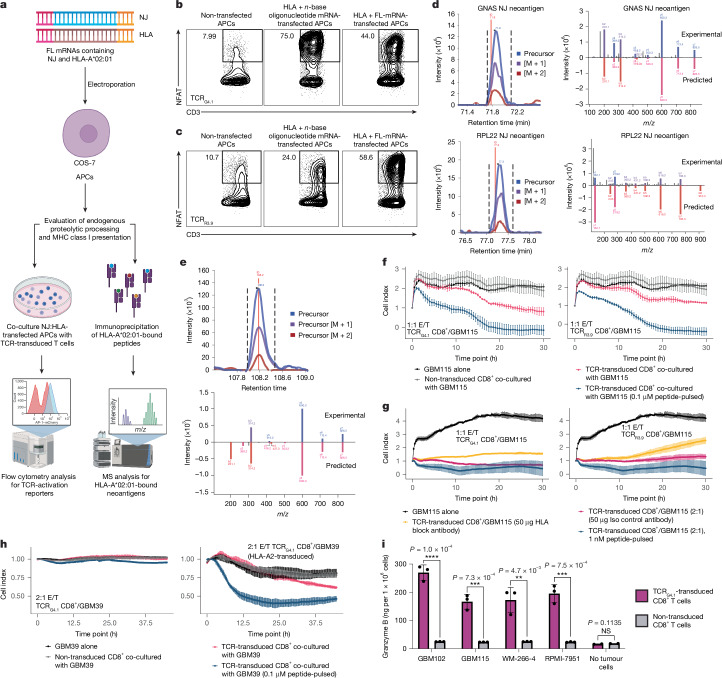
T cell-based immunotherapies hold promise in treating cancer by leveraging the immune system's recognition of cancer-specific antigens1. However, their efficacy is limited in tumours with few somatic mutations and substantial intratumoural heterogeneity2-4. Here we introduce a previously uncharacterized class of tumour-wide public neoantigens originating from RNA splicing aberrations in diverse cancer types. We identified T cell receptor clones capable of recognizing and targeting neoantigens derived from aberrant splicing in GNAS and RPL22. In cases with multi-site biopsies, we detected the tumour-wide expression of the GNAS neojunction in glioma, mesothelioma, prostate cancer and liver cancer. These neoantigens are endogenously generated and presented by tumour cells under physiologic conditions and are sufficient to trigger cancer cell eradication by neoantigen-specific CD8+ T cells. Moreover, our study highlights a role for dysregulated splicing factor expression in specific cancer types, leading to recurrent patterns of neojunction upregulation. These findings establish a molecular basis for T cell-based immunotherapies addressing the challenges of intratumoural heterogeneity.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: C.A.K. and I.E. are inventors on patents related to public neoantigen-specific TCRs unrelated to the present manuscript and are recipients of licensing revenue shared according to Memorial Sloan Kettering Cancer Center institutional policies. C.A.K. has consulted for or is on the scientific advisory boards for Achilles Therapeutics, Affini-T Therapeutics, Aleta BioTherapeutics, Bellicum Pharmaceuticals, Bristol Myers Squibb, Catamaran Bio, Cell Design Labs, Decheng Capital, G1 Therapeutics, Klus Pharma, Obsidian Therapeutics, PACT Pharma, Roche/Genentech, Royalty Pharma and T-knife. C.A.K. is a scientific co-founder and equity holder in Affini-T Therapeutics.
Figures












Update of
-
Tumor-wide RNA splicing aberrations generate immunogenic public neoantigens.bioRxiv [Preprint]. 2023 Oct 20:2023.10.19.563178. doi: 10.1101/2023.10.19.563178. bioRxiv. 2023. Update in: Nature. 2025 Mar;639(8054):463-473. doi: 10.1038/s41586-024-08552-0. PMID: 37904942 Free PMC article. Updated. Preprint.
References
-
- Russo, M. et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science366, 1473–1480 (2019). - PubMed
MeSH terms
Substances
Grants and funding
- T32 CA151022/CA/NCI NIH HHS/United States
- R01 CA269733/CA/NCI NIH HHS/United States
- P50 CA097257/CA/NCI NIH HHS/United States
- P50 CA217694/CA/NCI NIH HHS/United States
- T32 GM008568/GM/NIGMS NIH HHS/United States
- R01 CA222965/CA/NCI NIH HHS/United States
- R35 NS105068/NS/NINDS NIH HHS/United States
- P01 CA118816/CA/NCI NIH HHS/United States
- S10 OD028511/OD/NIH HHS/United States
- R01 CA286507/CA/NCI NIH HHS/United States
- R50 CA274229/CA/NCI NIH HHS/United States
- R37 CA259177/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
