

This is a preprint.
LRRC8A-containing anion channels promote glioblastoma proliferation via a WNK1/mTORC2-dependent mechanism
- PMID: 39975357
- PMCID: PMC11838495
- DOI: 10.1101/2025.02.02.636139
LRRC8A-containing anion channels promote glioblastoma proliferation via a WNK1/mTORC2-dependent mechanism
Abstract
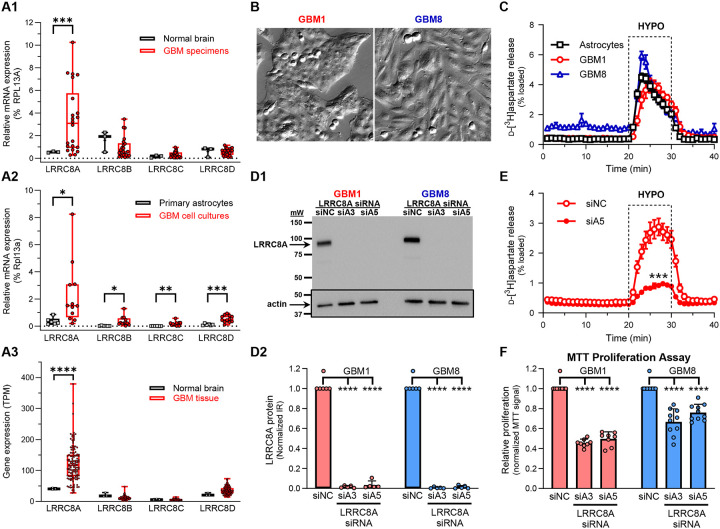
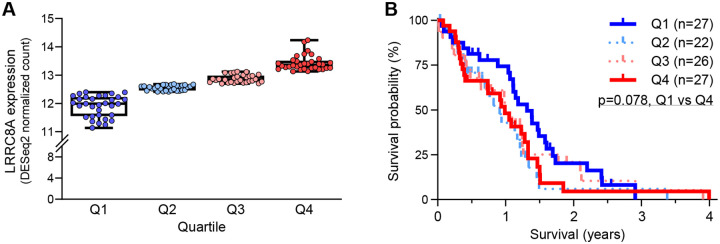
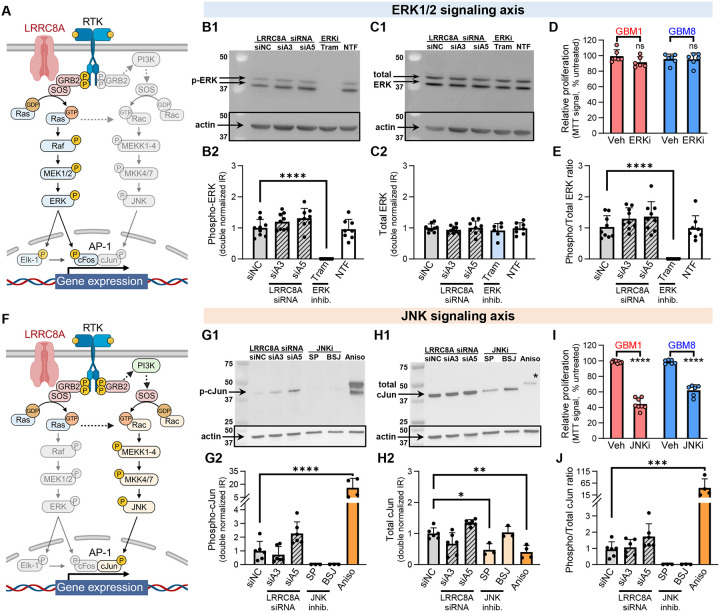
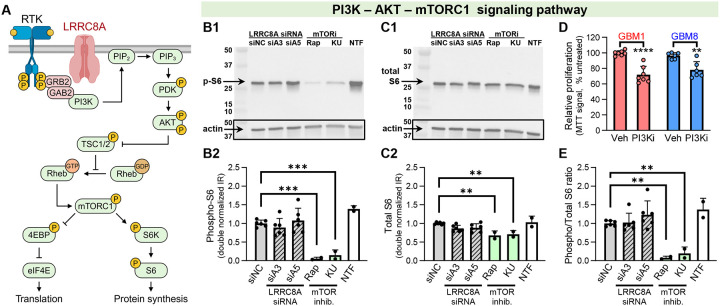
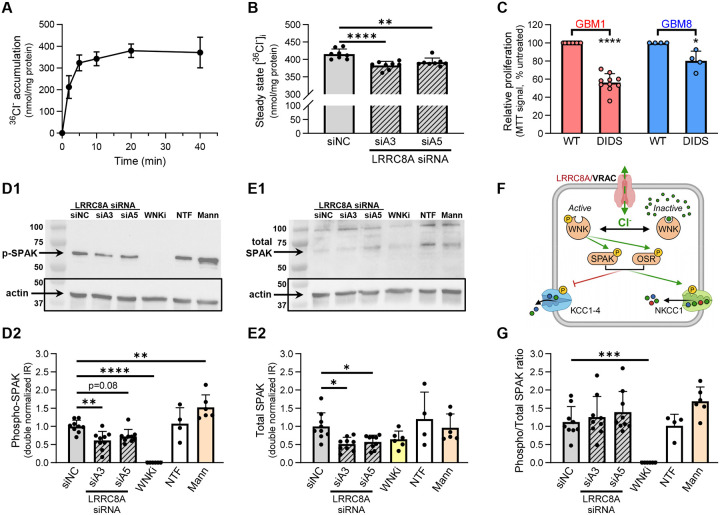
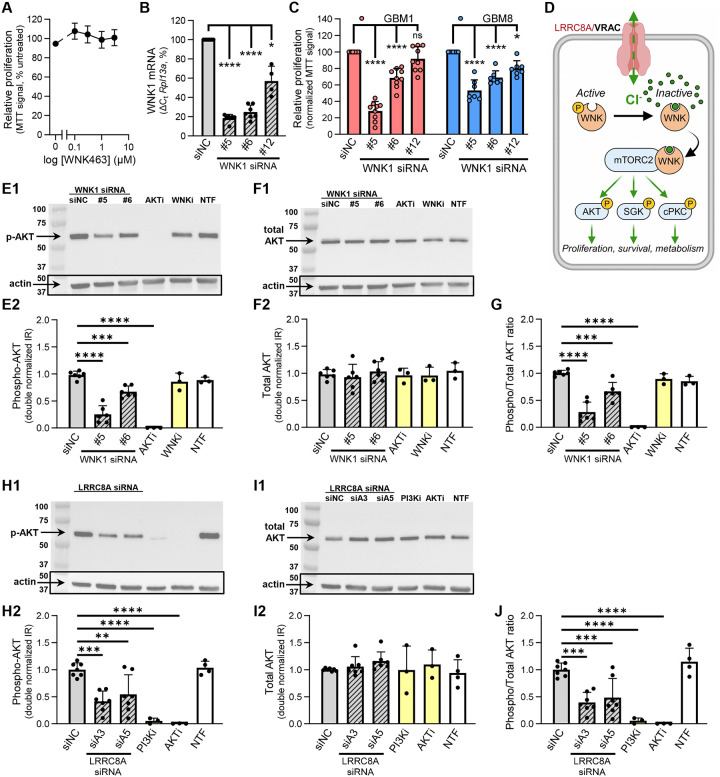
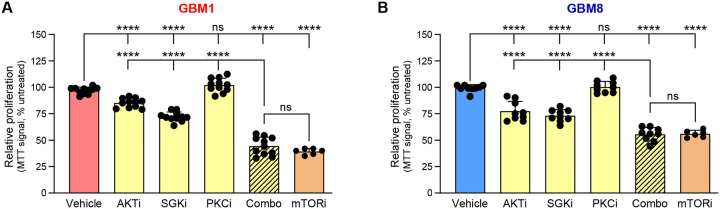
Leucine-rich repeat-containing protein 8A (LRRC8A) is the essential subunit of ubiquitous volume-regulated anion channels (VRACs). LRCC8A is overexpressed in several cancers and promotes negative survival outcomes via a poorly defined mechanism. Here, we explored the role of LRRC8A and VRACs in the progression of glioblastoma (GBM), the most common and deadly primary brain tumor. We found that, as compared to healthy controls, LRRC8A mRNA was strongly upregulated in surgical GBM specimens, patient-derived GBM cell lines, and GBM datasets from The Cancer Genome Atlas (TCGA). Our in-silico analysis indicated that patients belonging to the lowest LRRC8A expression quartile demonstrated a trend for extended life expectancy. In patient-derived GBM cultures, siRNA-driven LRRC8A knockdown reduced cell proliferation and additionally decreased intracellular chloride levels and inhibited activity of mTOR complex 2. The antiproliferative effect of LRRC8A downregulation was recapitulated with a pharmacological inhibitor of VRAC. Our ensuing biochemical and molecular biology analyses established that the LRRC8A-containing VRACs facilitate GBM proliferation via a new mechanism involving non-enzymatic actions of the chloride-sensitive protein kinase WNK1. Accordingly, the chloride-bound WNK1 stimulates mTORC2 and the mTORC2-dependent protein kinases AKT and SGK, which promote proliferation. These findings establish the new mTORC2-centric axis for VRAC dependent regulation of cellular functions and uncover potential targets for GBM intervention.
Keywords: Cancer; Cell biology; Molecular biology.
Conflict of interest statement
DECLARATION OF INTERESTS The authors report no competing interests.
Figures

References
-
- Wen P.Y. and Kesari S. (2008). Malignant gliomas in adults. N. Engl. J. Med. 359, 492507. - PubMed
-
- Brennan C.W., Verhaak R.G., McKenna A., Campos B., Noushmehr H., Salama S.R., Zheng S., Chakravarty D., Sanborn J.Z., Berman S.H., Beroukhim R., Bernard B., Wu C.J., Genovese G., Shmulevich I., Barnholtz-Sloan J., Zou L., Vegesna R., Shukla S.A., Ciriello G., Yung W.K., Zhang W., Sougnez C., Mikkelsen T., Aldape K., Bigner D.D., van Meir E.G., Prados M., Sloan A., Black K.L., Eschbacher J., Finocchiaro G., Friedman W., Andrews D.W., Guha A., Iacocca M., O’Neill B.P., Foltz G., Myers J., Weisenberger D.J., Penny R., Kucherlapati R., Perou C.M., Hayes D.N., Gibbs R., Marra M., Mills G.B., Lander E., Spellman P., Wilson R., Sander C., Weinstein J., Meyerson M., Gabriel S., Laird P.W., Haussler D., Getz G., and Chin L. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462477. - PMC - PubMed
-
- Cloughesy T.F., Cavenee W.K., and Mischel P.S. (2014). Glioblastoma: from molecular pathology to targeted treatment. Annu. Rev. Pathol. 9, 1–25. - PubMed
-
- Tan A.C., Ashley D.M., Lopez G.Y., Malinzak M., Friedman H.S., and Khasraw M. (2020). Management of glioblastoma: State of the art and future directions. CA Cancer J. Clin. 70, 299–312. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous