

This is a preprint.
Benchmarking, detection, and genotyping of structural variants in a population of whole-genome assemblies using the SVGAP pipeline
- PMID: 39975360
- PMCID: PMC11839052
- DOI: 10.1101/2025.02.07.637096
Benchmarking, detection, and genotyping of structural variants in a population of whole-genome assemblies using the SVGAP pipeline
Abstract
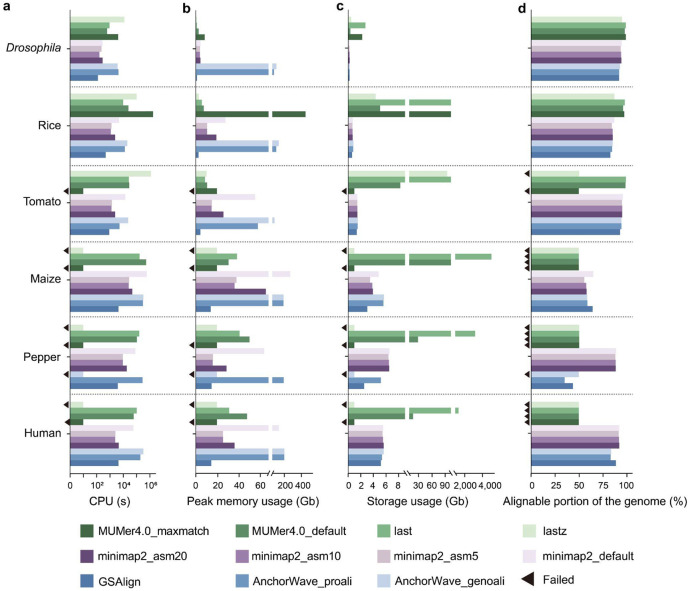
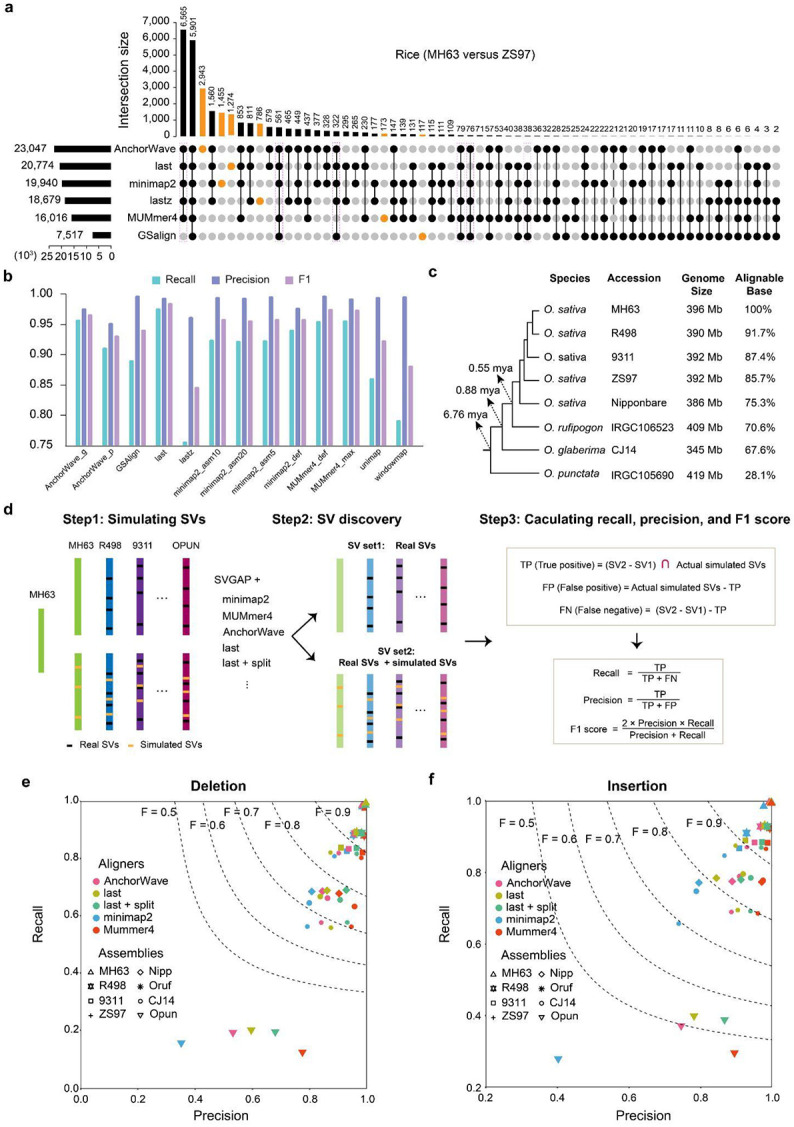
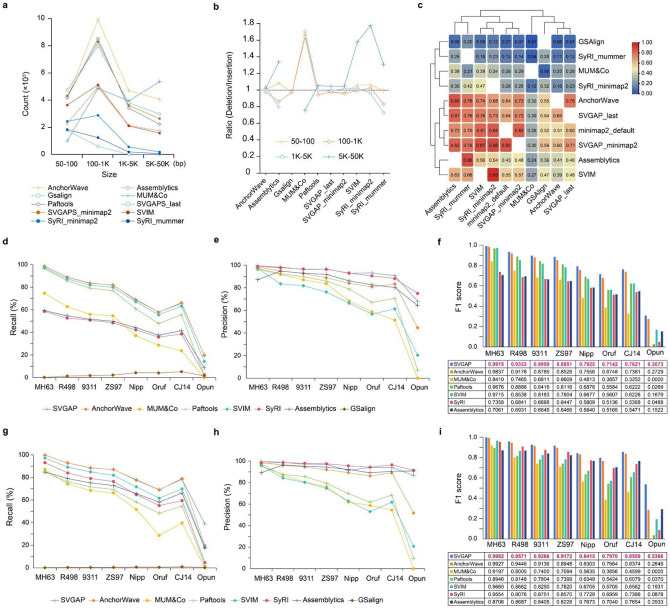
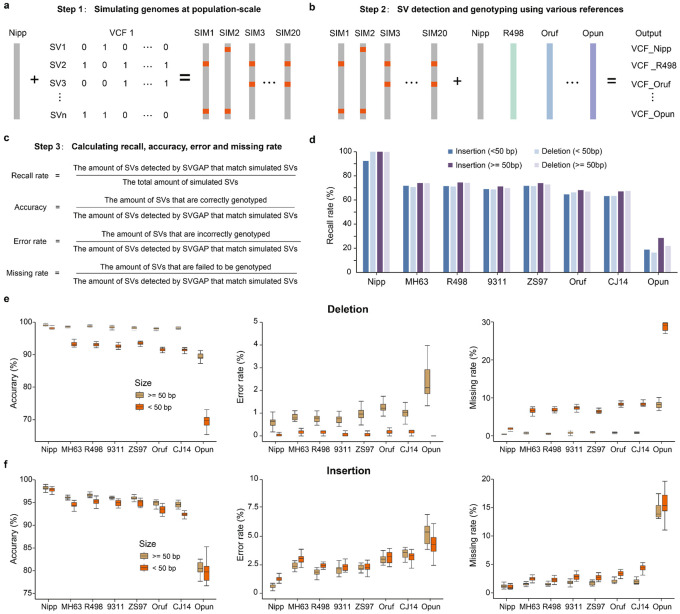
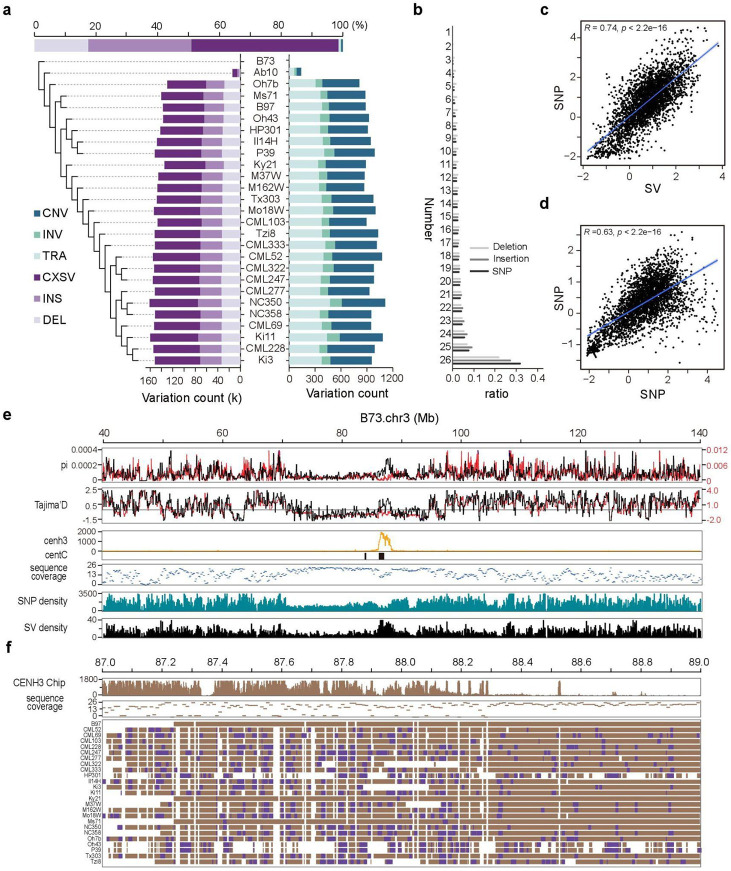
Comparisons of complete genome assemblies offer a direct procedure for characterizing all genetic differences among them. However, existing tools are often limited to specific aligners or optimized for specific organisms, narrowing their applicability, particularly for large and repetitive plant genomes. Here, we introduce SVGAP, a pipeline for structural variant (SV) discovery, genotyping, and annotation from high-quality genome assemblies at the population level. Through extensive benchmarks using simulated SV datasets at individual, population, and phylogenetic contexts, we demonstrate that SVGAP performs favorably relative to existing tools in SV discovery. Additionally, SVGAP is one of the few tools to address the challenge of genotyping SVs within large assembled genome samples, and it generates fully genotyped VCF files. Applying SVGAP to 26 maize genomes revealed hidden genomic diversity in centromeres, driven by abundant insertions of centromere-specific LTR-retrotransposons. The output of SVGAP is well-suited for pan-genome construction and facilitates the interpretation of previously unexplored genomic regions.
Figures

Similar articles
-
Accurate, scalable structural variant genotyping in complex genomes at population scales.Mol Biol Evol. 2025 Jul 29:msaf180. doi: 10.1093/molbev/msaf180. Online ahead of print. Mol Biol Evol. 2025. PMID: 40721218
-
VISTA: an integrated framework for structural variant discovery.Brief Bioinform. 2024 Jul 25;25(5):bbae462. doi: 10.1093/bib/bbae462. Brief Bioinform. 2024. PMID: 39297879 Free PMC article.
-
Tools for annotation and comparison of structural variation.F1000Res. 2017 Oct 3;6:1795. doi: 10.12688/f1000research.12516.1. eCollection 2017. F1000Res. 2017. PMID: 29123647 Free PMC article.
-
The challenges and importance of structural variation detection in livestock.Front Genet. 2014 Feb 18;5:37. doi: 10.3389/fgene.2014.00037. eCollection 2014. Front Genet. 2014. PMID: 24600474 Free PMC article. Review.
-
Genome-wide functional annotation of variants: a systematic review of state-of-the-art tools, techniques and resources.Front Pharmacol. 2025 Mar 3;16:1474026. doi: 10.3389/fphar.2025.1474026. eCollection 2025. Front Pharmacol. 2025. PMID: 40098614 Free PMC article. Review.
References
-
- Gaut B.S., Seymour D.K., Liu Q. and Zhou Y. (2018) Demography and its effects on genomic variation in crop domestication. Nat Plants, 4, 512–520. - PubMed
-
- Escaramís G., Docampo E. and Rabionet R. (2015) A decade of structural variants: description, history and methods to detect structural variation. Brief. Funct. Genomics, 14, 305–314. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources