

Inactivation of necroptosis-promoting protein MLKL creates a therapeutic vulnerability in colorectal cancer cells
- PMID: 39979285
- PMCID: PMC11842741
- DOI: 10.1038/s41419-025-07436-z
Inactivation of necroptosis-promoting protein MLKL creates a therapeutic vulnerability in colorectal cancer cells
Abstract
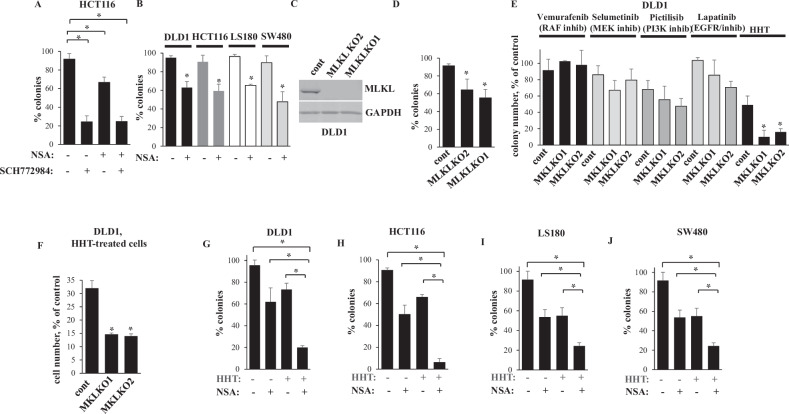
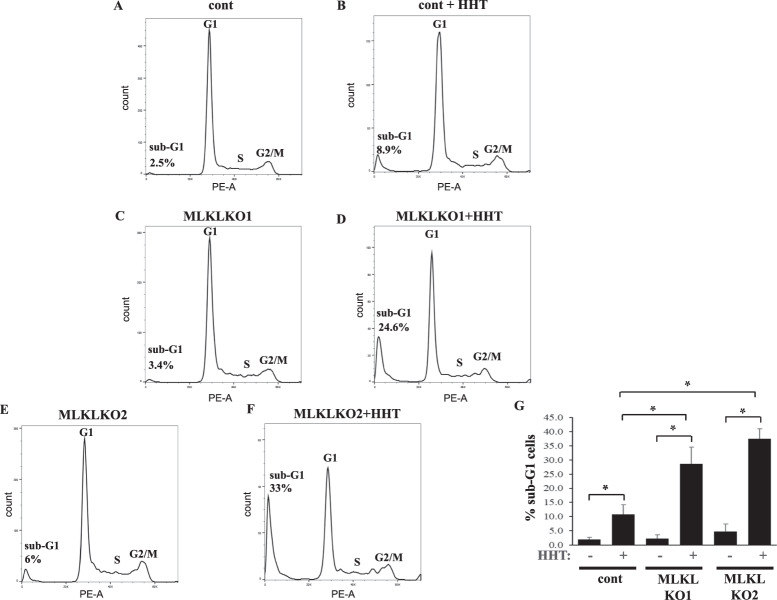
Mortality from colorectal cancer (CRC) is significant, and novel CRC therapies are needed. A pseudokinase MLKL typically executes necroptotic cell death, and MLKL inactivation protects cells from such death. However, we found unexpectedly that MLKL gene knockout enhanced CRC cell death caused by a protein synthesis inhibitor homoharringtonine used for chronic myeloid leukemia treatment. In an effort to explain this finding, we observed that MLKL gene knockout reduces the basal CRC cell autophagy and renders such autophagy critically dependent on the presence of VPS37A, a component of the ESCRT-I complex. We further found that the reason why homoharringtonine enhances CRC cell death caused by MLKL gene knockout is that homoharringtonine activates p38 MAP kinase and thereby prevents VPS37A from supporting autophagy in MLKL-deficient cells. We observed that the resulting inhibition of the basal autophagy in CRC cells triggers their parthanatos, a cell death type driven by poly(ADP-ribose) polymerase hyperactivation. Finally, we discovered that a pharmacological MLKL inhibitor necrosulfonamide strongly cooperates with homoharringtonine in suppressing CRC cell tumorigenicity in mice. Thus, while MLKL promotes cell death during necroptosis, MLKL supports the basal autophagy in CRC cells and thereby protects them from death. MLKL inactivation reduces such autophagy and renders the cells sensitive to autophagy inhibitors, such as homoharringtonine. Hence, MLKL inhibition creates a therapeutic vulnerability that could be utilized for CRC treatment.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethics approval and consent to participate: Animal studies were approved by Dalhousie University Committee on Laboratory Animals, protocol 20-081.
Figures







References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
