

Galactosialidosis: A Report of Three Cases Diagnosed With a Founder Genetic Mutation in the Bahraini Population
- PMID: 39981487
- PMCID: PMC11840274
- DOI: 10.7759/cureus.77750
Galactosialidosis: A Report of Three Cases Diagnosed With a Founder Genetic Mutation in the Bahraini Population
Abstract
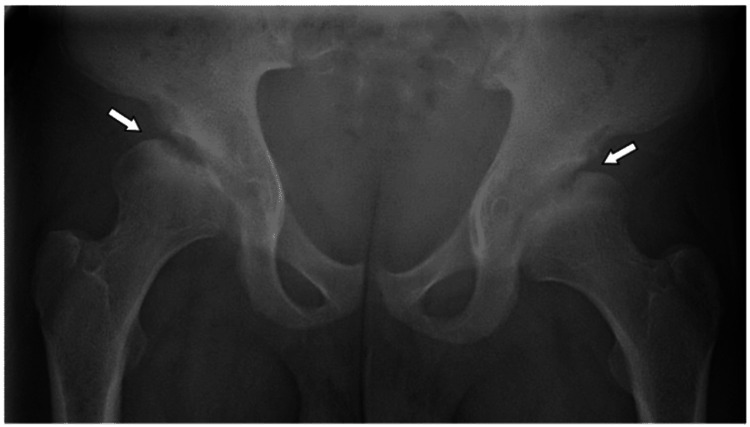
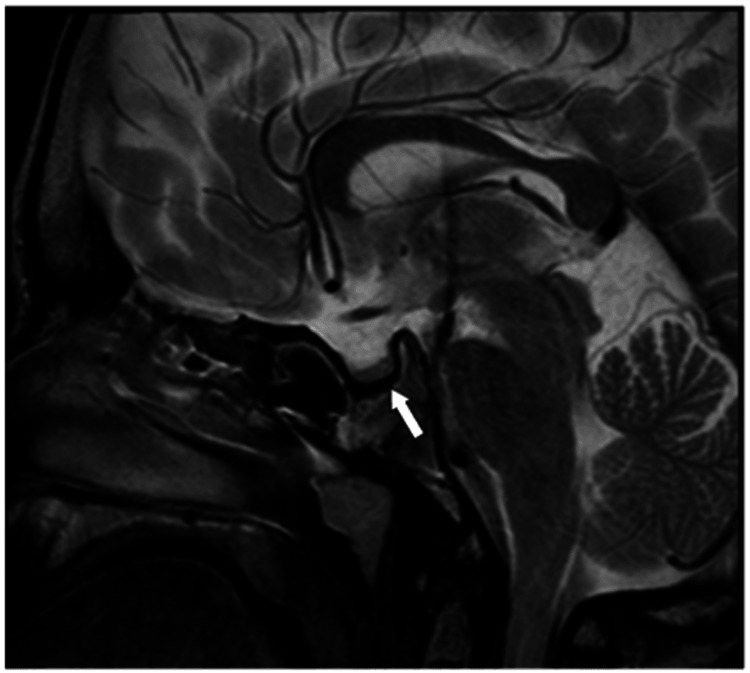
Galactosialidosis (GS, OMIM #256540) is a rare metabolic disorder resulting from mutations in the protective protein/cathepsin A (PPCA) or CTSA gene, which is characterized by malfunction of the lysosomal glycoprotein degradation and subsequent intra-lysosomal accumulation of sialyloligosaccharides and glycopeptides. It follows an autosomal recessive inheritance pattern. This systemic disease is characterized by typical clinical features such as short stature, coarse facial features, vertebral deformities, gastrointestinal manifestations, particularly hepatosplenomegaly, cardiac abnormalities, hearing loss, and macular cherry-red spots. GS is classified into three subtypes based on the age of onset and presenting symptoms. The three types include the early infantile (EI) form, which is the most severe; the late infantile form; and the juvenile/adult form. Here, we present three newly diagnosed cases of late-infantile GS in Bahraini patients, all sharing the same previously reported homozygous mutation in the CTSA gene (c.607C>A, p.Pro203Thr), confirmed by targeted mutation analysis. This mutation has been identified in nine Bahraini patients, reflecting a founder effect in the Bahraini population. All three patients presented with coarse facial features, short stature, and poor vision, alongside skeletal deformities. Patient 1 had significant bilateral hip osteoarthritis, while Patient 2. showed lumbar lordosis and extensive bilateral hip avascular necrosis. Patient 3 presented with thoracolumbar levoscoliosis and kyphoscoliosis. Additionally, in Patient 1 and Patient 2 cardiac manifestations were noted, including valvular heart disease. Patient 3 had mild left ventricular hypertrophy (LVH), aortic regurgitation, and mitral regurgitation, along with diffuse angiokeratomas. All patients are currently receiving supportive care and management. This case report highlights the importance of early diagnosis and multidisciplinary care of patients with GS.
Keywords: bahraini; founder mutation; galactosialidosis; lysosomal lipid storage disease; novel mutation.
Copyright © 2025, Alsahlawi et al.
Conflict of interest statement
Human subjects: Consent for treatment and open access publication was obtained or waived by all participants in this study. Conflicts of interest: In compliance with the ICMJE uniform disclosure form, all authors declare the following: Payment/services info: All authors have declared that no financial support was received from any organization for the submitted work. Financial relationships: All authors have declared that they have no financial relationships at present or within the previous three years with any organizations that might have an interest in the submitted work. Other relationships: All authors have declared that there are no other relationships or activities that could appear to have influenced the submitted work.
Figures





References
-
- d’Azzo A, Andria G, Bonten E, Annunziata I. The Online Metabolic and Molecular Bases of Inherited Disease. New York: The McGraw-Hill Education; 2019. Galactosialidosis.
-
- Beta-galactosidase deficiency in young adults. Wenger DA, Goodman SI, Myers GG. Lancet. 1974;2:1319–1320. - PubMed
-
- Macular cherry-red spots and myoclonus with dementia-coexistent neuraminidase and beta-galactosidase deficiencies. Wenger DA, Tarby TJ, Wharton C. Biochem Biophys Res Commun. 1978;82:589–595. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous