

Linear-Scaling Local Natural Orbital-Based Full Triples Treatment in Coupled-Cluster Theory
- PMID: 39981711
- PMCID: PMC11912218
- DOI: 10.1021/acs.jctc.4c01716
Linear-Scaling Local Natural Orbital-Based Full Triples Treatment in Coupled-Cluster Theory
Abstract
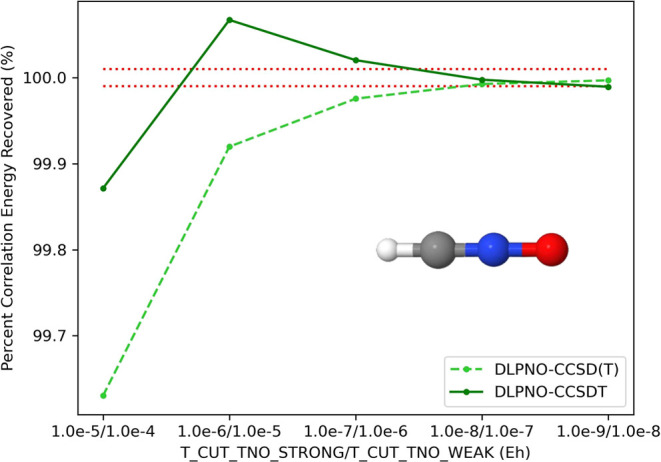
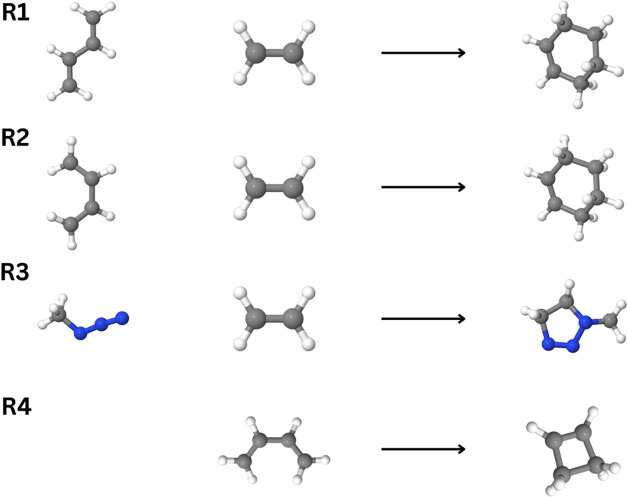
We present an efficient, asymptotically linear-scaling implementation of the canonically coupled-cluster method with singles, doubles, and full triples excitations (CCSDT) method. We apply the domain-based local pair natural orbital (DLPNO) approach for computing CCSDT amplitudes. Our method, called DLPNO-CCSDT, uses the converged coupled-cluster amplitudes from a preceding DLPNO-CCSD(T) computation as a starting point for the solution of the CCSDT equations in the local natural orbital basis. To simplify the working equations, we t1-dress our two-electron integrals and Fock matrices, allowing our equations to take on the form of CCDT. With appropriate parameters, our method can recover more than 99.99% of the total canonical CCSDT correlation energy. In addition, we demonstrate that our method consistently yields sub-kJ mol-1 errors in relative energies when compared to canonical CCSDT, and, likewise, when computing the difference between CCSDT and CCSD(T). Finally, to highlight the low scaling of our algorithm, we present timings on linear alkanes (up to 30 carbons and 730 basis functions) and water clusters (up to 131 water molecules and 3144 basis functions).
Conflict of interest statement
The authors declare no competing financial interest.
Figures




References
-
- Crawford T. D.; Schaefer H. F. An introduction to coupled cluster theory for computational chemists. Rev. Comp. Chem. 2000, 14, 33–136. 10.1002/9780470125915.ch2. - DOI
-
- Bartlett R. J.; Musial M. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 2007, 79, 29110.1103/RevModPhys.79.291. - DOI
-
- Sherrill C. D.; Schaefer H. F. The Configuration Interaction Method: Advances in Highly Correlated Approaches. Adv. Quantum Chem. 1999, 34, 143–269. 10.1016/s0065-3276(08)60532-8. - DOI
-
- Cramer C. J.Essentials of Computational Chemistry: Theories and Models. In Theoretical Chemistry Accounts; Wiley, 2002.
-
- Stanton J. F. Why CCSD(T) works: a different perspective. Chem. Phys. Lett. 1997, 281, 130–134. 10.1016/S0009-2614(97)01144-5. - DOI
LinkOut - more resources
Full Text Sources