

Activation of CAMK2 by pseudokinase PEAK1 represents a targetable pathway in triple negative breast cancer
- PMID: 39984440
- PMCID: PMC11845518
- DOI: 10.1038/s41467-025-57046-8
Activation of CAMK2 by pseudokinase PEAK1 represents a targetable pathway in triple negative breast cancer
Abstract
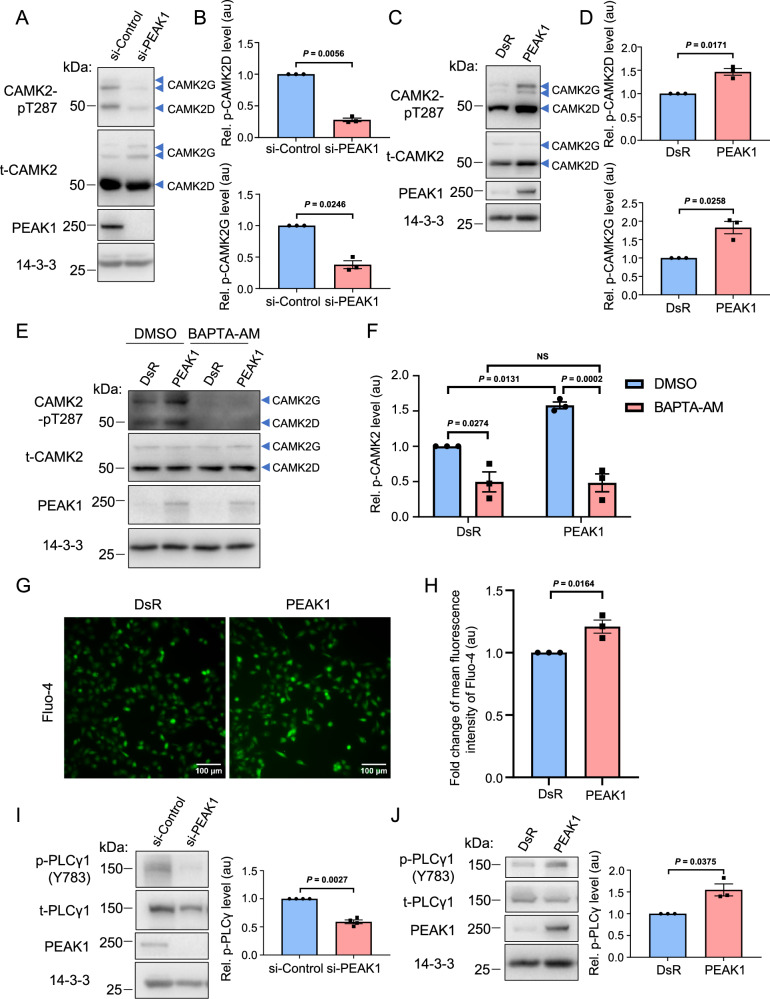
The PEAK family of pseudokinases, comprising PEAK1-3, play oncogenic roles in several poor prognosis human cancers, including triple negative breast cancer (TNBC). However, therapeutic targeting of pseudokinases is challenging due to their lack of catalytic activity. To address this, we screen for PEAK1 effectors and identify calcium/calmodulin-dependent protein kinase 2 (CAMK2)D and CAMK2G. PEAK1 promotes CAMK2 activation in TNBC cells via PLCγ1/Ca2+ signalling and direct binding to CAMK2. In turn, CAMK2 phosphorylates PEAK1 to enhance association with PEAK2, which is critical for PEAK1 oncogenic signalling. To achieve pharmacologic targeting of PEAK1/CAMK2, we repurpose RA306, a second generation CAMK2 inhibitor. RA306 inhibits PEAK1-enhanced migration and invasion of TNBC cells in vitro and significantly attenuates TNBC xenograft growth and metastasis in a manner mirrored by PEAK1 ablation. Overall, these studies establish PEAK1 as a critical cell signalling nexus that integrates Ca2+ and tyrosine kinase signals and identify CAMK2 as a therapeutically 'actionable' target downstream of PEAK1.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures







Update of
-
Feed-forward stimulation of CAMK2 by the oncogenic pseudokinase PEAK1 generates a therapeutically "actionable" signalling axis in triple negative breast cancer.bioRxiv [Preprint]. 2024 Nov 22:2024.02.14.580406. doi: 10.1101/2024.02.14.580406. bioRxiv. 2024. Update in: Nat Commun. 2025 Feb 22;16(1):1871. doi: 10.1038/s41467-025-57046-8. PMID: 38405732 Free PMC article. Updated. Preprint.
References
-
- Reiterer, V., Eyers, P. A. & Farhan, H. Day of the dead: pseudokinases and pseudophosphatases in physiology and disease. Trends Cell Biol.24, 489–505 (2014). - PubMed
-
- Hou, J. et al. Distinct PEAK3 interactors and outputs expand the signaling potential of the PEAK pseudokinase family. Sci. Signal15, eabj3554 (2022). - PubMed
-
- Patel, O., Roy, M. J., Murphy, J. M. & Lucet, I. S. The PEAK family of pseudokinases, their role in cell signalling and cancer. FEBS J.287, 4183–4197 (2020). - PubMed
MeSH terms
Substances
Grants and funding
- T32 GM139789/GM/NIGMS NIH HHS/United States
- National Research Service Award T32 GM139789/UMASS | University of Massachusetts Amherst (UMass Amherst)
- MCRF21036/Victorian Cancer Agency (VCA)
- NIH R35GM145376/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- DE240100992/Department of Education and Training | Australian Research Council (ARC)
- DP220103638/Department of Education and Training | Australian Research Council (ARC)
- Ideas Grant 2003599/Department of Health | National Health and Medical Research Council (NHMRC)
- R35 GM145376/GM/NIGMS NIH HHS/United States
- DP190103672/Department of Education and Training | Australian Research Council (ARC)
- NBCF Fellowship IIRS-20-032/National Breast Cancer Foundation (NBCF)
LinkOut - more resources
Full Text Sources
Miscellaneous
