

KBG syndrome: report and follow-up on three unrelated patients observed at different ages
- PMID: 39985057
- PMCID: PMC11846256
- DOI: 10.1186/s13052-025-01884-1
KBG syndrome: report and follow-up on three unrelated patients observed at different ages
Abstract
Background: KBG syndrome (MIM #148050) is a rare genetic disease, showing an autosomal dominant pattern of inheritance. It was first described by Herrmann et al. in 1975 in three affected families, whose initial letters gave origin to the acronym. A peculiar facies including triangular face, synophrys, macrodontia of the upper central incisors, as well as short stature, skeletal defects and neurodevelopmental disorders (developmental delay, intellectual disability, epilepsy) are the main features of the syndrome. Mutations of the ankirin repeat domain 11 gene (ANKRD11), which harbors at chromosome 16q24.3, have been associated to the syndrome. The encoded protein inhibits ligand-dependent activation of transcription. Due to the growing number of detected ANKRD11 variants associated to phenotypes with various degree of severity, the precise definition of the clinical and genomic profiles of patients is important, also in the perspective of a better understanding of the molecular bases of the disease, genotype-phenotype correlation, and management of affected subjects.
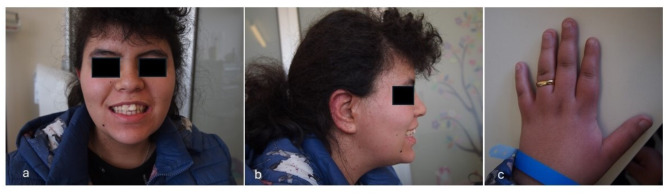
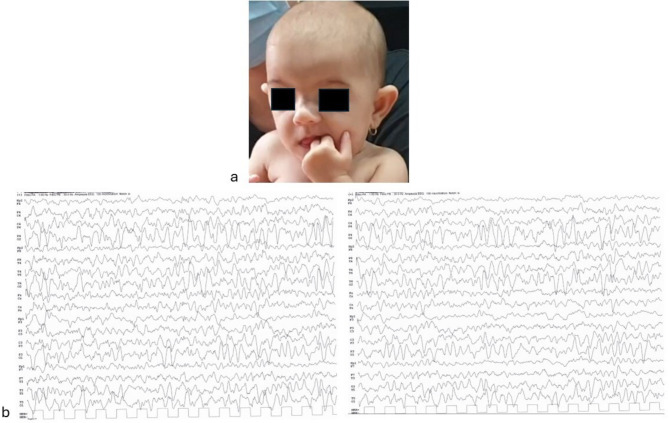
Cases presentation: We report on three unrelated patients, observed in as many different Italian (Sicily, Veneto and Friuli-Venezia-Giulia regions) Pediatric Neurology and Medical Genetics outpatient services, showing variously present typical dysmorphic features (e.g., triangular face, macrodontia of upper incisors, brachydactyly), growth retardation and impaired neurodevelopmental profiles (i.e. developmental delay, EEG abnormalities/epilepsy) compatible with KBG syndrome diagnosis. In Patient 1, next generation sequencing analysis of a panel of genes involved in developmental delay and autism spectrum disorders detected two mutations, a pathogenic heterozygous frameshift variant of the ANKRD11 gene (already described in the literature), and a heterozygous missense one in EHMT1 (previously reported as well, and associated with Kleefstra syndrome); in Patient 2, array comparative genomic hybridization (a-CGH) analysis identified a 634 Kb 16q24.3-24.3 deletion involving several genes (CDT1, APRT, GALNS, TRAPPC2L, ACSF3, CDH15), besides ANKRD11, some of which are related with developmental disorders. Finally in Patient 3, Sanger sequencing of the ANKRD11 gene, performed due to the specific diagnostic suspicion raised for precocious teething observed at age 3 months, evidenced an intragenic deletion allowing thus an early diagnosis of disease.
Conclusions: We underline similarities and differences among our patients, and their specific genetic and clinical features, in addition to the variable diagnostic tests used for the diagnosis, reached at different developmental age, i.e. infancy, childhood and adolescence. Pediatricians must be aware of KBG syndrome and should be able, as well, to raise the diagnostic suspicion, especially in the presence of peculiar dysmorphic features, short stature, developmental delay, intellectual disability and epilepsy. Prompt diagnosis may allow to better address any associated emerging neuropsychological and behavioral issues improving the quality of life of the patient and the whole family.
Keywords: ANKRD11 gene; 16q24.3 deletion; Next generation sequencing; Sanger sequencing; array-CGH.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Written informed consent was obtained from parents at admission of their children. The study was approved by the Mother and Child Department of the University of Palermo, ethics committee Palermo 1 (Palermo, Italy). All procedures performed in this report were in accordance with the ethical standards of the institutional and national research committee, and with the 1964 Helsinki declaration and its later amendments, or comparable ethical standards. Consent for publication: Written informed consent was obtained from patient’s parents for publication of this case report and accompanying images. Competing interests: We declare that Giovanni Corsello and Gregorio Serra, last and first author of the present manuscript, are respectively Editor-in-Chief and Associate Editor of Italian Journal of Pediatrics.
Figures


References
-
- Nardello R, Mangano GD, Antona V, Fontana A, Striano P, Giorgio E, Brusco A, Mangano S, Salpietro V. Electroclinical features and outcome of ANKRD11-related KBG syndrome: a novel report and literature review. Seizure - Eur J Epilepsy. 2021;85:151–4. - PubMed
-
- Low K, Ashraf T, Canham N, Clayton-Smith J, Deshpande C, Donaldson A, Fisher R, Flinter F, Foulds N, Fryer A, Gibson K, Hayes I, Hills A, Holder S, Irving M, Joss S, Kivuva E, Lachlan K, Magee A, McConnell V, McEntagart M, Metcalfe K, Montgomery T, Newbury-Ecob R, Stewart F, Turnpenny P, Vogt J, Fitzpatrick D, Williams M. DDD Study; Smithson S. Clinical and genetic aspects of KBG syndrome. Am J Med Genet A. 2016;170(11):2835–46. - PMC - PubMed
-
- Herrmann J, Pallister PD, Tiddy W, Opitz JM. The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig Artic Ser. 1975;11(5):7–18. - PubMed
-
- Sirmaci A, Spiliopoulos M, Brancati F, Powell E, Duman D, Abrams A, Bademci G, Agolini E, Guo S, Konuk B, Kavaz A, Blanton S, Digilio MC, Dallapiccola B, Young J, Zuchner S, Tekin M. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am J Hum Genet. 2011.12;89(2):289– 94. - PMC - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
