

Distinct outcomes from targeted perturbations of the multi-subunit SCFSkp2 E3 ubiquitin ligase in blocking Trp53/Rb1-null prostate tumorigenesis
- PMID: 39987265
- PMCID: PMC11846996
- DOI: 10.1038/s42003-025-07662-3
Distinct outcomes from targeted perturbations of the multi-subunit SCFSkp2 E3 ubiquitin ligase in blocking Trp53/Rb1-null prostate tumorigenesis
Abstract
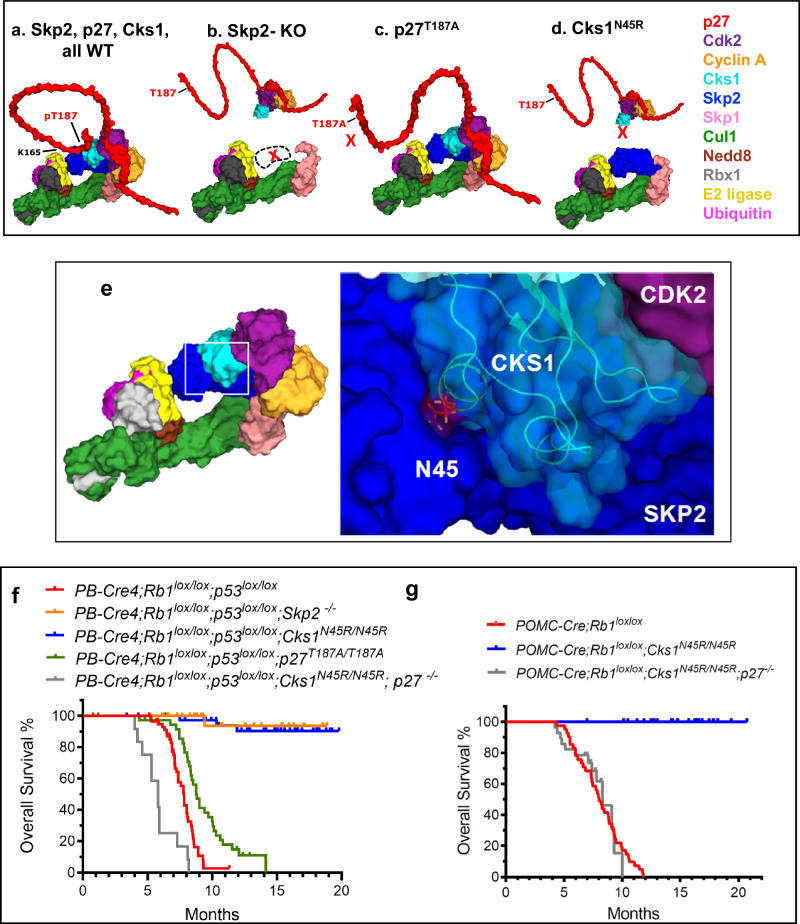
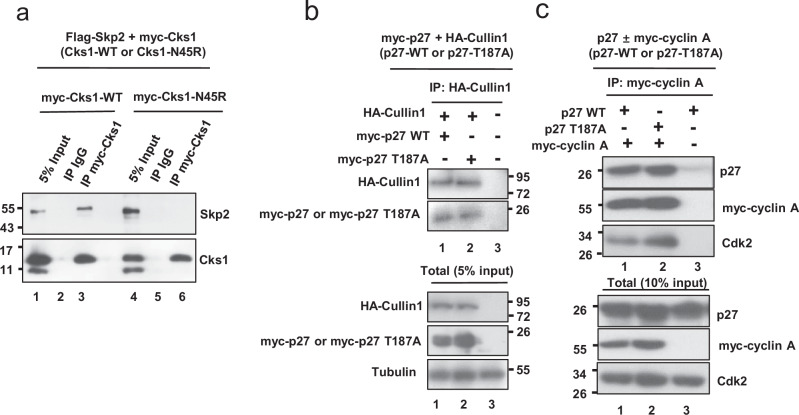
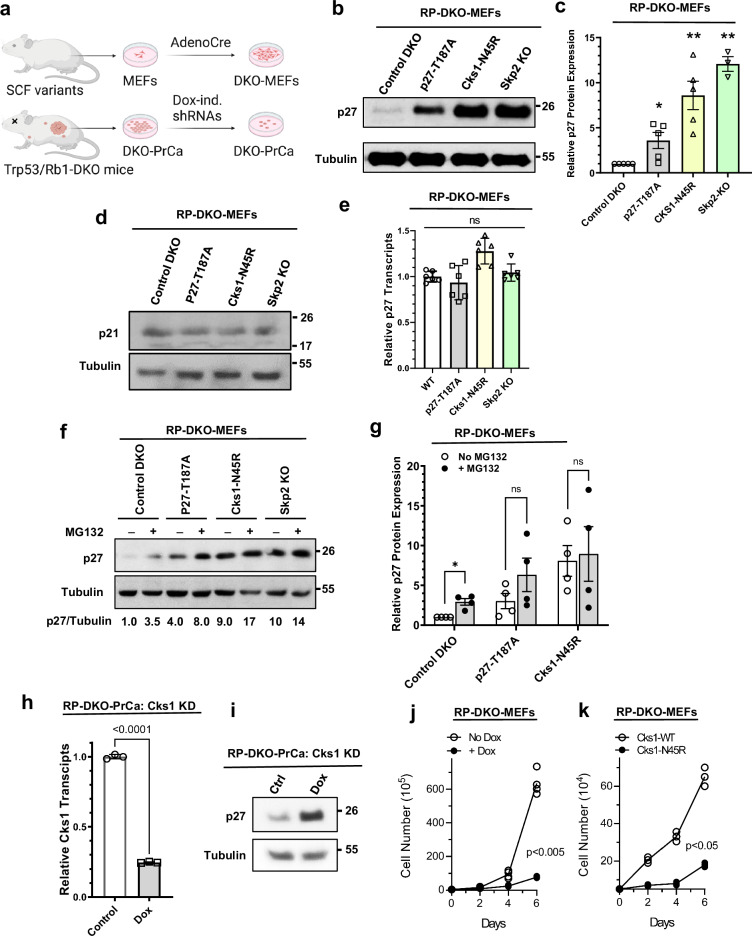
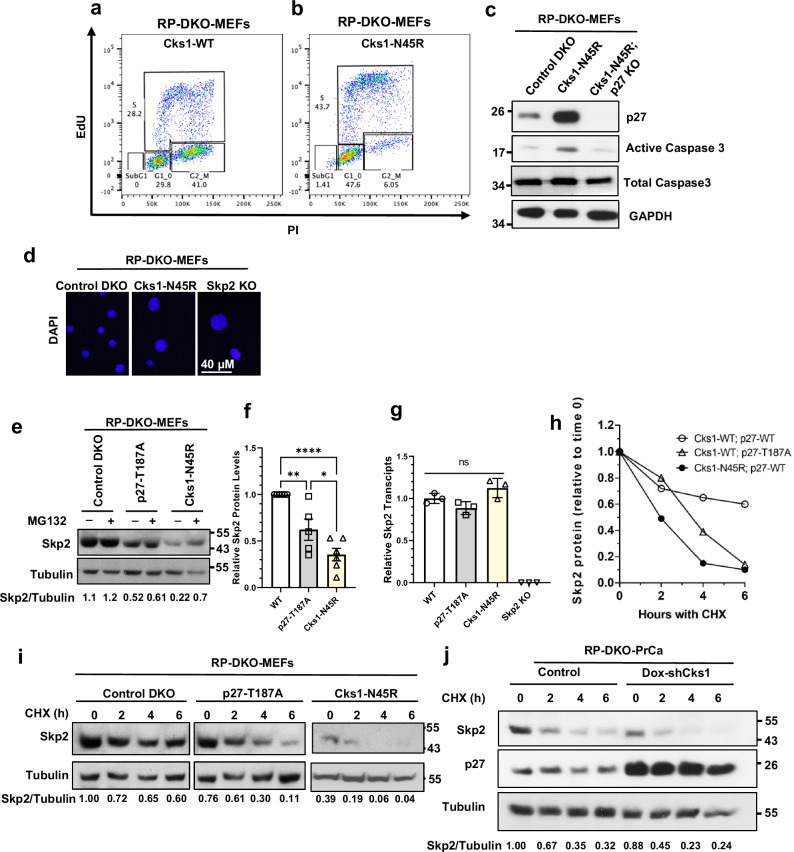
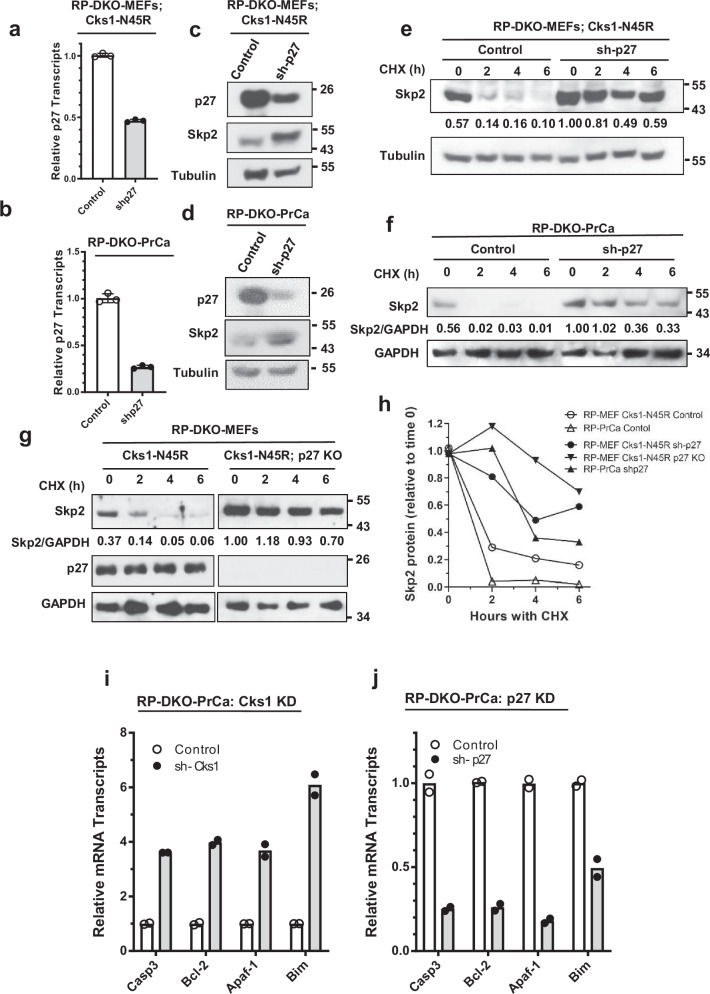
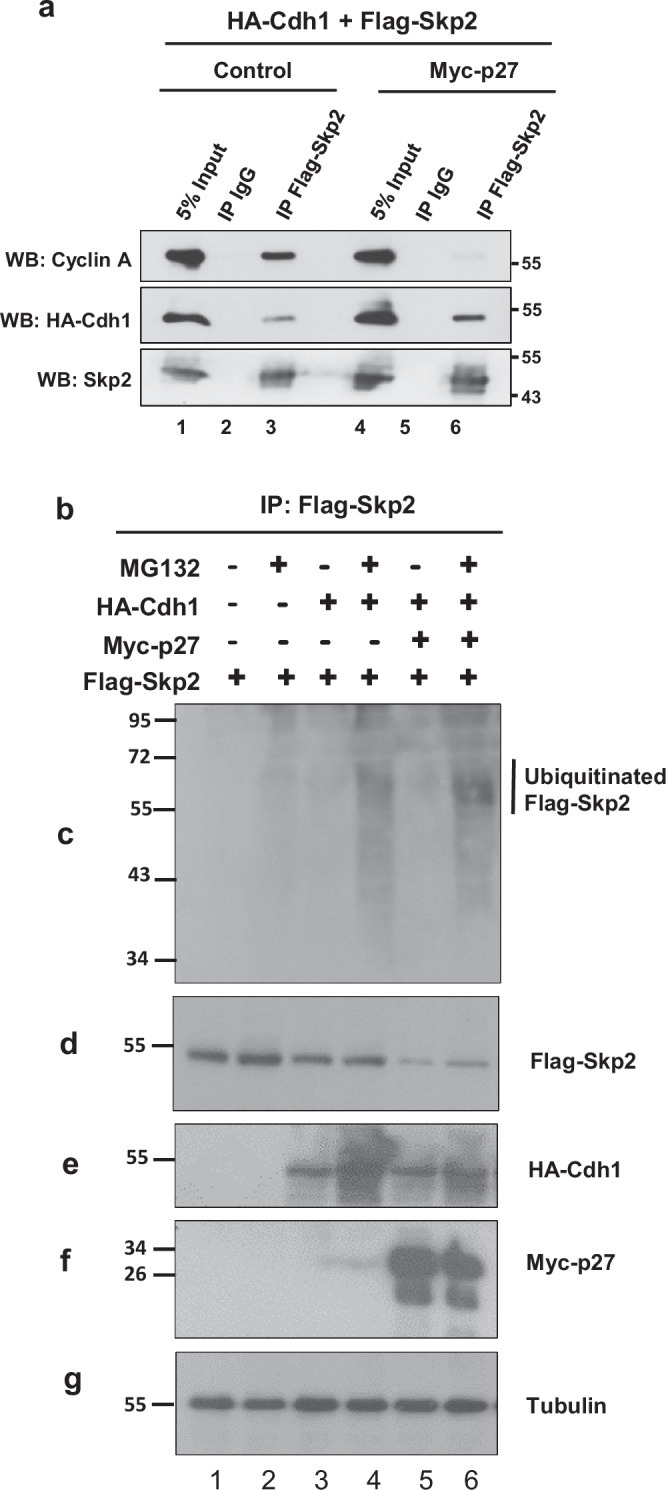
Identifying effective therapies targeting multi-protein complexes that lack catalytic sites or cofactor pockets remains a long-standing challenge. The proto-oncogene, ubiquitin E3 ligase SCFSkp2, is one such target. SCFSkp2 promotes the proteasomal degradation of the cyclin-dependent kinase inhibitor p27, which controls cell cycle progression. Targeted knockout of Rb1/Trp53 causes metastatic prostate cancer in mice; additional knockout of Skp2 completely blocks tumorigenesis. We compared gene-edited mice that carried two different single amino acid changes in the SCFSkp2 complex, structurally predicted to inhibit the degradation of p27. Mutation of the SCFSkp2 accessory protein Cks1 (Cks1N45R) completely blocked Rb1/Trp53-driven prostate tumorigenesis, phenocopying Skp2 knockout, whereas a mutation directly stabilizing p27 (p27T187A) did not. This was consistent with structural models that predicted the binding of both p27 and p27T187A to the SCFSkp2/Cks1/Cdk2/CyclinA/p27 complex, and their subsequent ubiquitination and degradation, albeit at different rates. Two binding modes, which differ in their dependence on phosphorylated T187, are predicted by the model. Studies confirmed the role of p27 in mediating tumorigenesis in Rb1/Trp53 mutant tumors and revealed a mutually destabilizing Skp2 and p27 feedback loop. The integration of gene editing, drug-surrogate mutations, and mouse tumor models offers a blueprint for studying SCFSkp2 and other multi-subunit biomedical targets.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare the following competing interests: E.S. is a consultant for Sandoz Pharmaceuticals. J.P.M. is a co-founder of Exo Therapeutics, a company focused on substrate-binding sites. The remaining authors declare no competing interests.
Figures

Similar articles
-
Targeted Inhibition of the E3 Ligase SCFSkp2/Cks1 Has Antitumor Activity in RB1-Deficient Human and Mouse Small-Cell Lung Cancer.Cancer Res. 2020 Jun 1;80(11):2355-2367. doi: 10.1158/0008-5472.CAN-19-2400. Epub 2020 Apr 7. Cancer Res. 2020. PMID: 32265224 Free PMC article.
-
p27T187A knockin identifies Skp2/Cks1 pocket inhibitors for advanced prostate cancer.Oncogene. 2017 Jan 5;36(1):60-70. doi: 10.1038/onc.2016.175. Epub 2016 May 16. Oncogene. 2017. PMID: 27181203 Free PMC article.
-
Inhibitors of SCF-Skp2/Cks1 E3 ligase block estrogen-induced growth stimulation and degradation of nuclear p27kip1: therapeutic potential for endometrial cancer.Endocrinology. 2013 Nov;154(11):4030-45. doi: 10.1210/en.2013-1757. Epub 2013 Sep 13. Endocrinology. 2013. PMID: 24035998 Free PMC article.
-
Targeting the untargetable: RB1-deficient tumours are vulnerable to Skp2 ubiquitin ligase inhibition.Br J Cancer. 2022 Oct;127(6):969-975. doi: 10.1038/s41416-022-01898-0. Epub 2022 Jun 25. Br J Cancer. 2022. PMID: 35752713 Free PMC article. Review.
-
The role of Skp2 and its substrate CDKN1B (p27) in colorectal cancer.J Gastrointestin Liver Dis. 2015 Jun;24(2):225-34. doi: 10.15403/jgld.2014.1121.242.skp2. J Gastrointestin Liver Dis. 2015. PMID: 26114183 Review.
References
-
- Nava Rodrigues, D. et al. RB1 heterogeneity in advanced metastatic castration-resistant prostate cancer. Clin. Cancer Res.25, 687–697 (2019). - PubMed
-
- Zhou, Z. et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res.66, 7889–7898 (2006). - PubMed
MeSH terms
Substances
Grants and funding
- RO1CA201458/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- S10 OD026852/OD/NIH HHS/United States
- R01 CA201458/CA/NCI NIH HHS/United States
- P30 CA013330/CA/NCI NIH HHS/United States
- P30CA13330/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
