

Emergence of inflammatory fibroblasts with aging in Hermansky-Pudlak syndrome associated pulmonary fibrosis
- PMID: 39987372
- PMCID: PMC11846979
- DOI: 10.1038/s42003-025-07589-9
Emergence of inflammatory fibroblasts with aging in Hermansky-Pudlak syndrome associated pulmonary fibrosis
Abstract
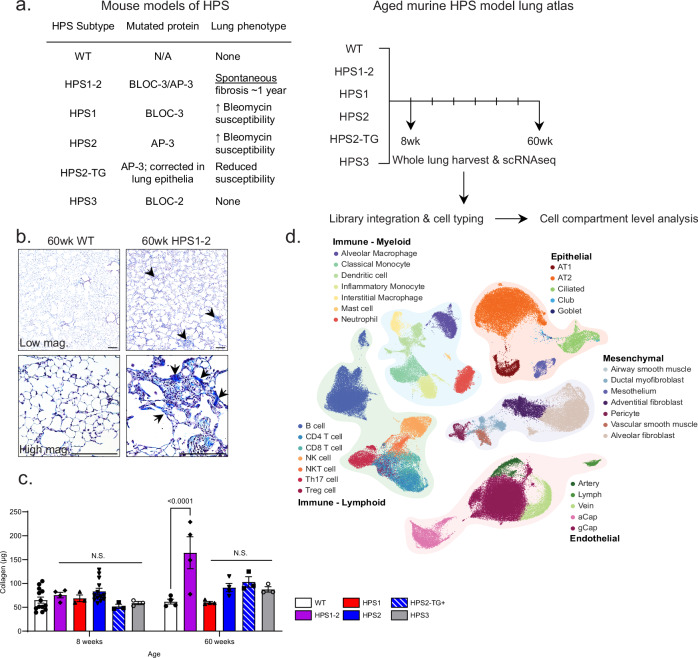
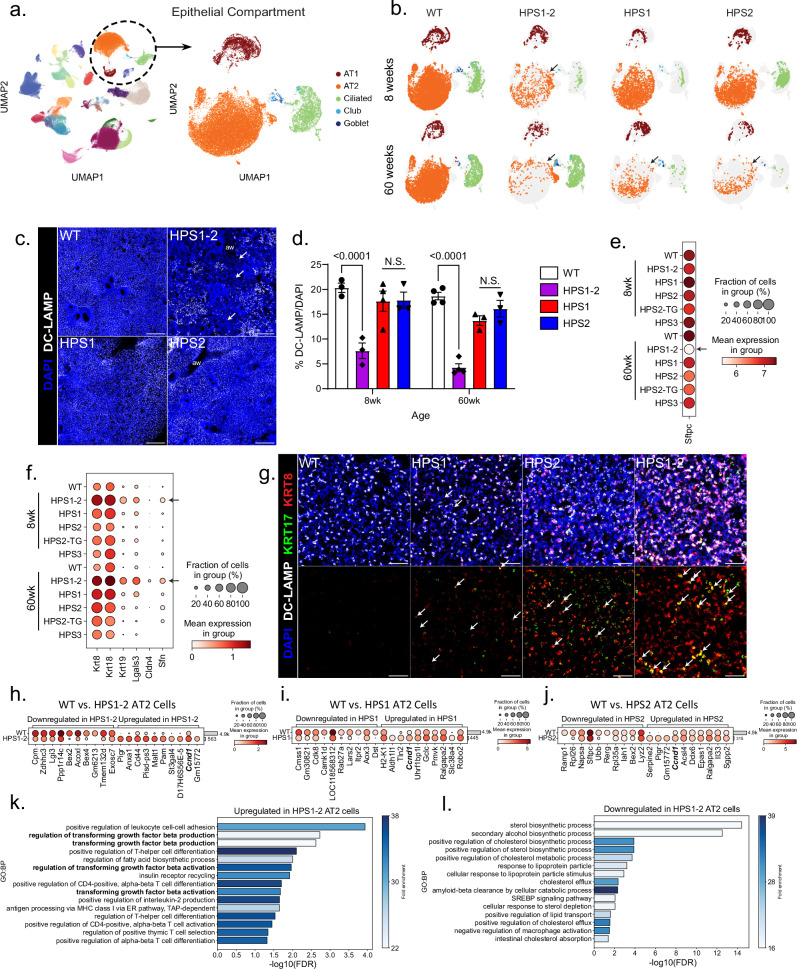
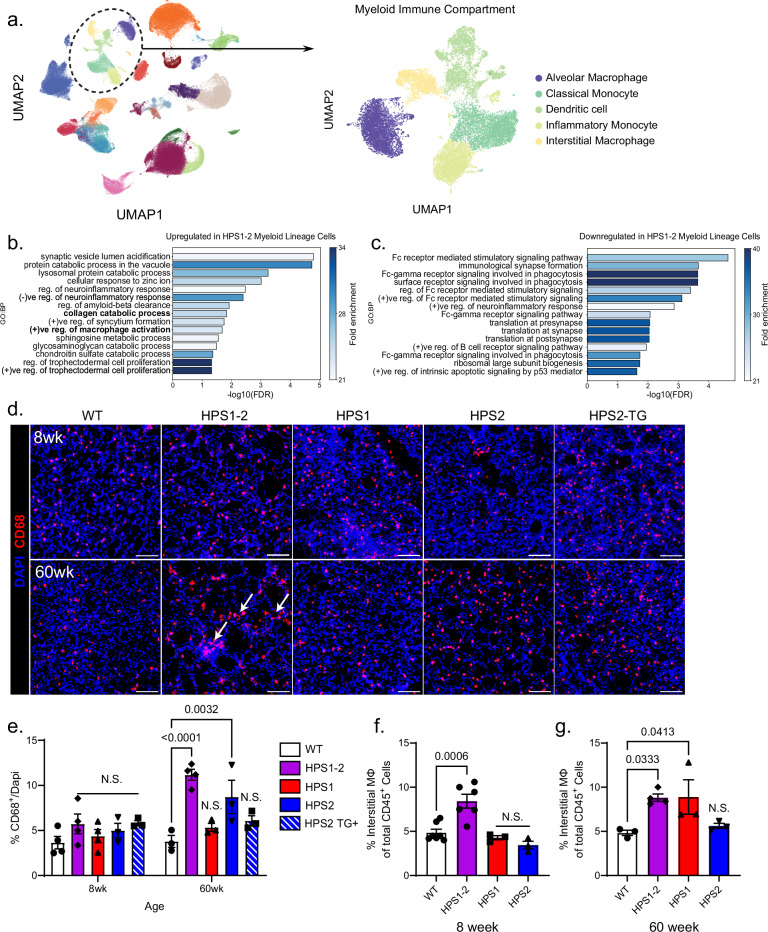
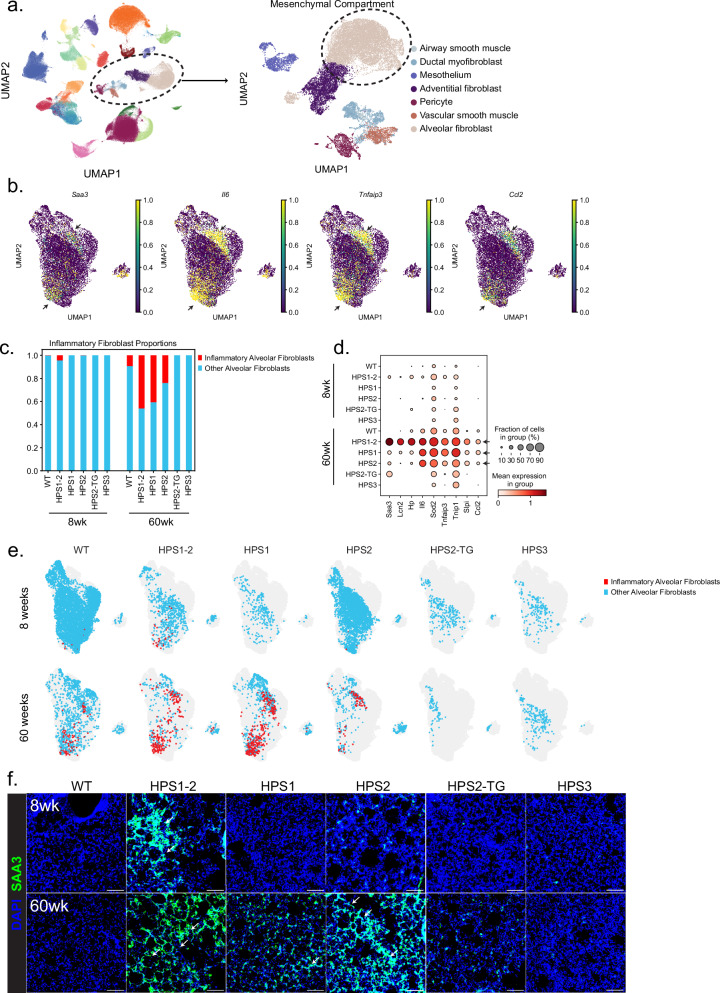
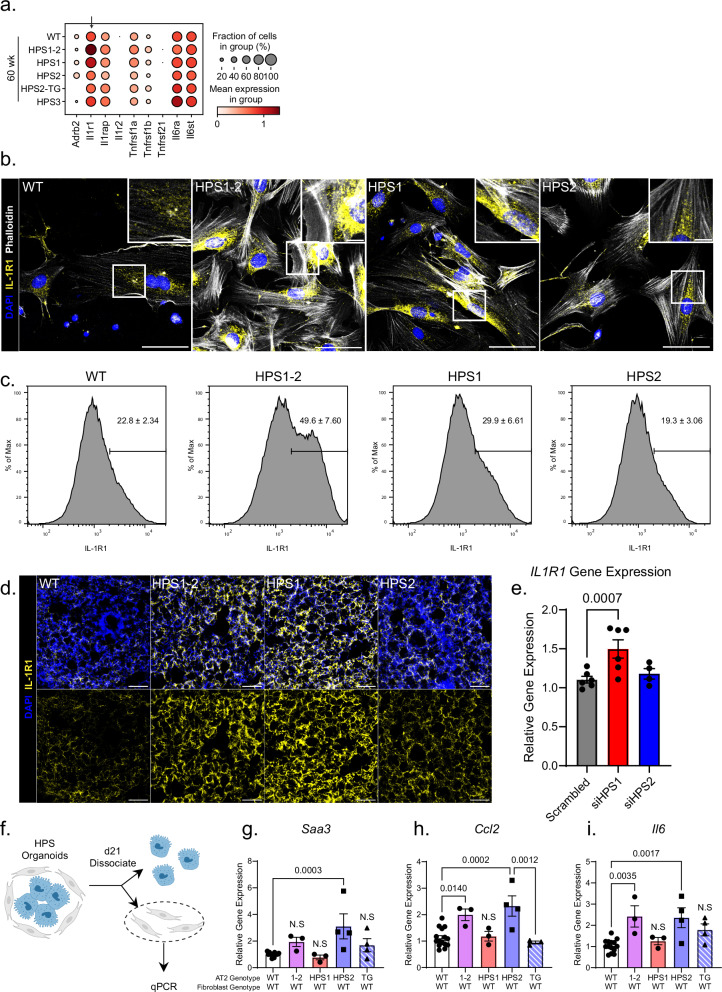
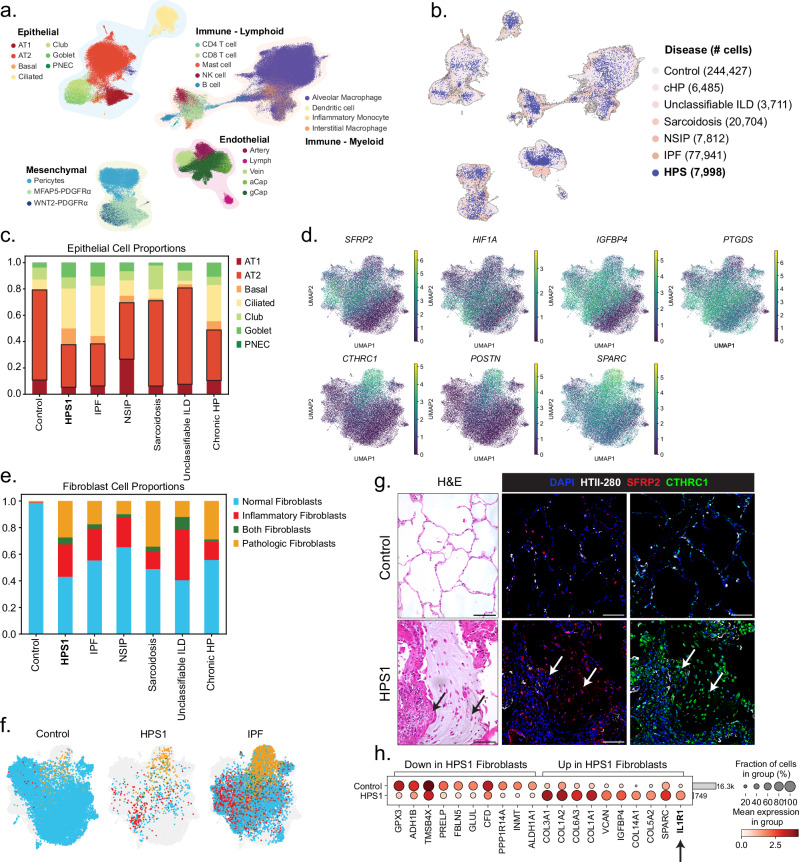
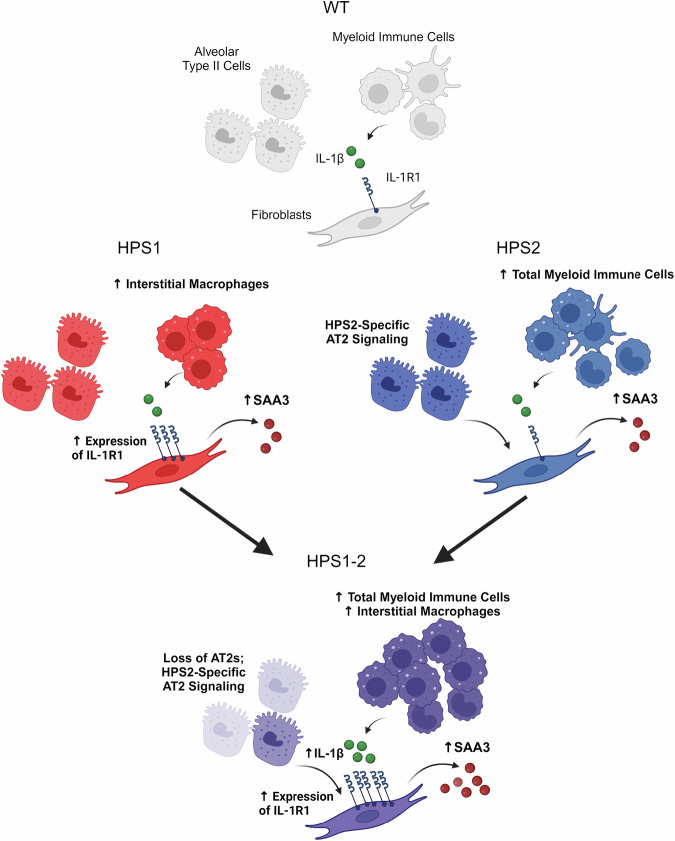
The longitudinal cellular interactions that drive pulmonary fibrosis are not well understood. To investigate the disease underpinnings associated with fibrosis onset and progression, we generated a scRNA-seq atlas of lungs from young and aged mouse models of multiple subtypes of Hermansky-Pudlak syndrome (HPS), a collection of rare autosomal recessive diseases associated with albinism, platelet dysfunction, and pulmonary fibrosis. We have identified an age-dependent increase in SAA3+ inflammatory lung fibroblasts in HPS mice, including in double-mutant HPS1-2 mice which develop spontaneous fibrosis. HPS1 fibroblasts show increased expression of IL-1R1, whereas alveolar type II epithelial cells from HPS2 mice induce the inflammatory gene signature in co-cultured fibroblasts. scRNA-seq of lung tissue from three HPS1 patients similarly shows the presence of inflammatory fibroblasts and increased IL1R1 expression on fibroblasts. These data posit complex interactions between dysfunctional epithelial cells, inflammatory fibroblasts, and recruited immune cells, suggesting potential opportunities for mitigation of the fibrotic cascade.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: B.J.B. reports equity in MannKind Corporation. JLT acknowledges grants to the institution from the National Institute of Health, AstraZeneca, Boehringer Ingelheim, and Care Dx; and advisory board participation for Avalyn and Sanofi. E.C. reports consulting fees from CSL Behring and United Therapeutics, and meeting travel support from Pulmocide Ltd. E.C. also reports board participation for the International Society of Heart and Lung Transplantation, regional representative role for the UNOS Lung Committee, and as a consultant for device group for the FDA. J.A.K. reports grants from Boehringer Ingelheim and scientific advisory board membership at APIE and ARDA. L.R.Y. reports scientific advisory board participation for Boehringer Ingelheim and Sanofi and royalties from Wolters Kluwer for UpToDate authorship. All conflicts of interest reported are outside the scope of the submitted work.
Figures
References
-
- Martinez, F. J. et al. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Prim. 3, 10.1038/nrdp.2017.74 (2017). - PubMed
MeSH terms
Grants and funding
- R01HL119503/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 HL145372/HL/NHLBI NIH HHS/United States
- K24 HL143281/HL/NHLBI NIH HHS/United States
- R01HL155821/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R35 GM146835/GM/NIGMS NIH HHS/United States
- P30 CA014236/CA/NCI NIH HHS/United States
- K08 HL140129/HL/NHLBI NIH HHS/United States
- R01 HL119503/HL/NHLBI NIH HHS/United States
- K24HL143281/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R35GM146835/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R00HL141684/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R00 HL141684/HL/NHLBI NIH HHS/United States
- K08 HL150226/HL/NHLBI NIH HHS/United States
- T32 HL007586/HL/NHLBI NIH HHS/United States
- R01 HL155821/HL/NHLBI NIH HHS/United States
- R56 HL167937/HL/NHLBI NIH HHS/United States
- R56HL167937/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
LinkOut - more resources
Full Text Sources
Medical
