

This is a preprint.
Systems-Level Interactome Mapping Reveals Actionable Protein Network Dysregulation Across the Alzheimer's Disease Spectrum
- PMID: 39989971
- PMCID: PMC11844643
- DOI: 10.21203/rs.3.rs-5930673/v1
Systems-Level Interactome Mapping Reveals Actionable Protein Network Dysregulation Across the Alzheimer's Disease Spectrum
Abstract
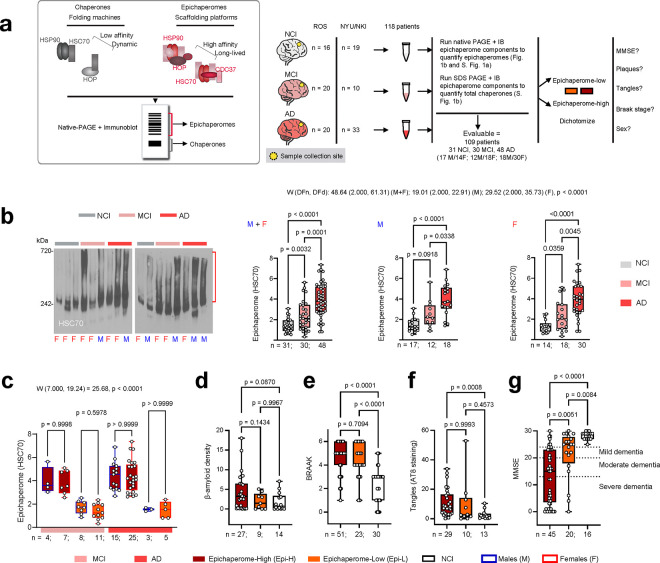
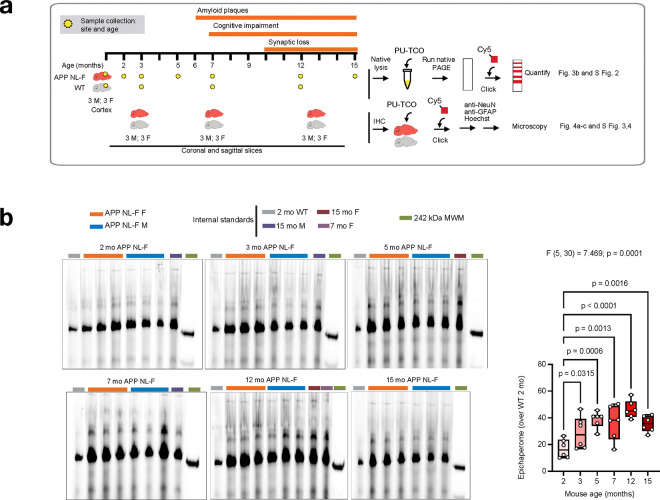
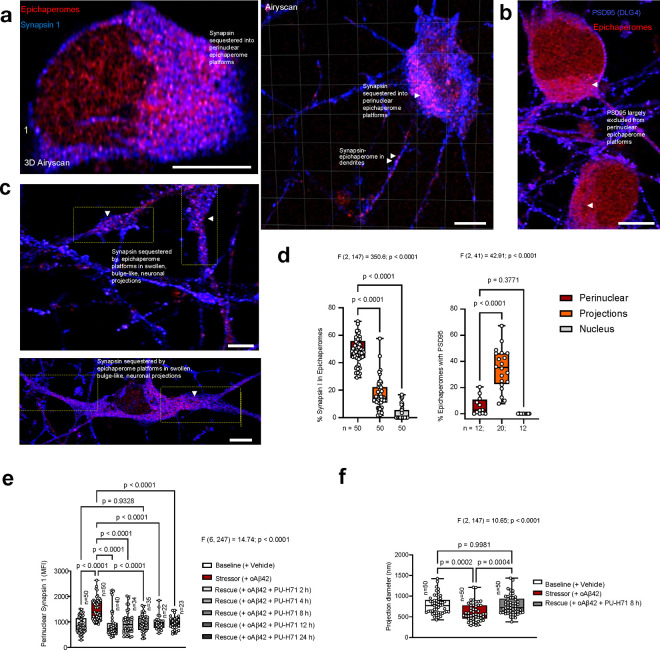
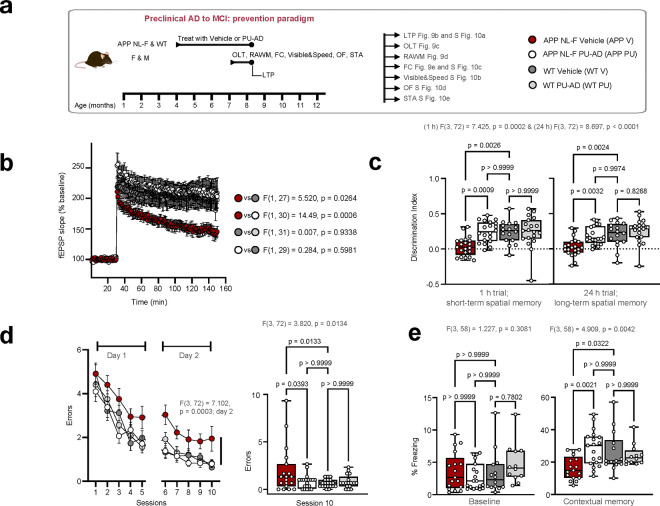
Alzheimer's disease (AD) progresses as a continuum, from preclinical stages to late-stage cognitive decline, yet the molecular mechanisms driving this progression remain poorly understood. Here, we provide a systems-level map of protein-protein interaction (PPI) network dysfunction across the AD spectrum and uncover epichaperomes-stable scaffolding platforms formed by chaperones and co-factors-as central drivers of this process. Using over 100 human brain specimens, mouse models, and human neurons, we show that epichaperomes emerge early, even in preclinical AD, and progressively disrupt multiple PPI networks critical for synaptic function and neuroplasticity. Glutamatergic neurons, essential for learning and memory, exhibit heightened vulnerability, with their dysfunction driven by protein sequestration into epichaperome scaffolds, independent of changes in protein expression. Notably, pharmacological disruption of epichaperomes with PU-AD restores PPI network integrity and reverses synaptic and cognitive deficits, directly linking epichaperome-driven network dysfunction to AD pathology. These findings establish epichaperomes as key mediators of molecular collapse in AD and identify network-centric intervention strategies as a promising avenue for disease-modifying therapies.
Conflict of interest statement
COMPETING INTERESTS Memorial Sloan Kettering Cancer Center holds the intellectual rights to the epichaperome portfolio. G.C., A.R. and S.S. are inventors on the licensed intellectual property. All other authors declare no competing interests.
Figures






References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
