

This is a preprint.
Nuclear regulatory disturbances precede and predict the development of Type-2 diabetes in Asian populations
- PMID: 39990582
- PMCID: PMC11844604
- DOI: 10.1101/2025.02.14.25322264
Nuclear regulatory disturbances precede and predict the development of Type-2 diabetes in Asian populations
Abstract
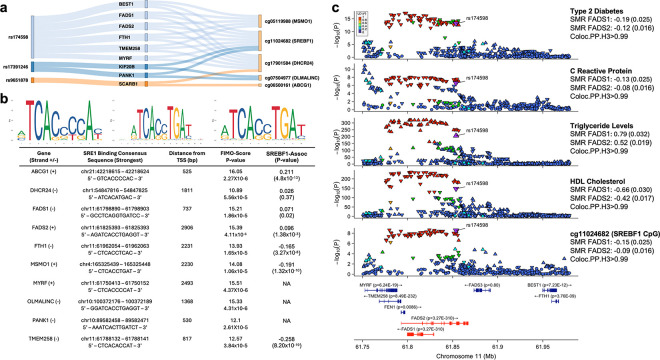
To identify biomarkers and pathways to Type-2 diabetes (T2D), a major global disease, we completed array-based epigenome-wide association in whole blood in 5,709 Asian people. We found 323 Sentinel CpGs (from 314 genetic loci) that predict future T2D. The CpGs reveal coherent, nuclear regulatory disturbances in canonical immune activation pathways, as well as metabolic networks involved in insulin signalling, fatty acid metabolism and lipid transport, which are causally linked to development of T2D. The CpGs have potential clinical utility as biomarkers. An array-based composite Methylation Risk Score (MRS) is predictive for future T2D (RR: 5.2 in Q4 vs Q1; P=7×10-25), and is additive to genetic risk. Targeted methylation sequencing revealed multiple additional CpGs predicting T2D, and synthesis of a sequencing-based MRS that is strongly predictive for T2D (RR: 8.3 in Q4 vs Q1; P=1.0×10-11). Importantly, MRS varies between Asian ethnic groups, in a way that explains a large fraction of the difference in T2D risk between populations. We thus provide new insights into the nuclear regulatory disturbances that precede development of T2D, and reveal the potential for sequence-based DNA methylation markers to inform risk stratification in diabetes prevention.
Figures















References
-
- Anjana R. M. et al. Metabolic non-communicable disease health report of India: the ICMR-INDIAB national cross-sectional study (ICMR-INDIAB-17). Lancet Diabetes Endocrinol. 11, 474–489 (2023). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources