

Multiscale Dissection of Spatial Heterogeneity by Integrating Multi-Slice Spatial and Single-Cell Transcriptomics
- PMID: 39999288
- PMCID: PMC12005799
- DOI: 10.1002/advs.202413124
Multiscale Dissection of Spatial Heterogeneity by Integrating Multi-Slice Spatial and Single-Cell Transcriptomics
Abstract
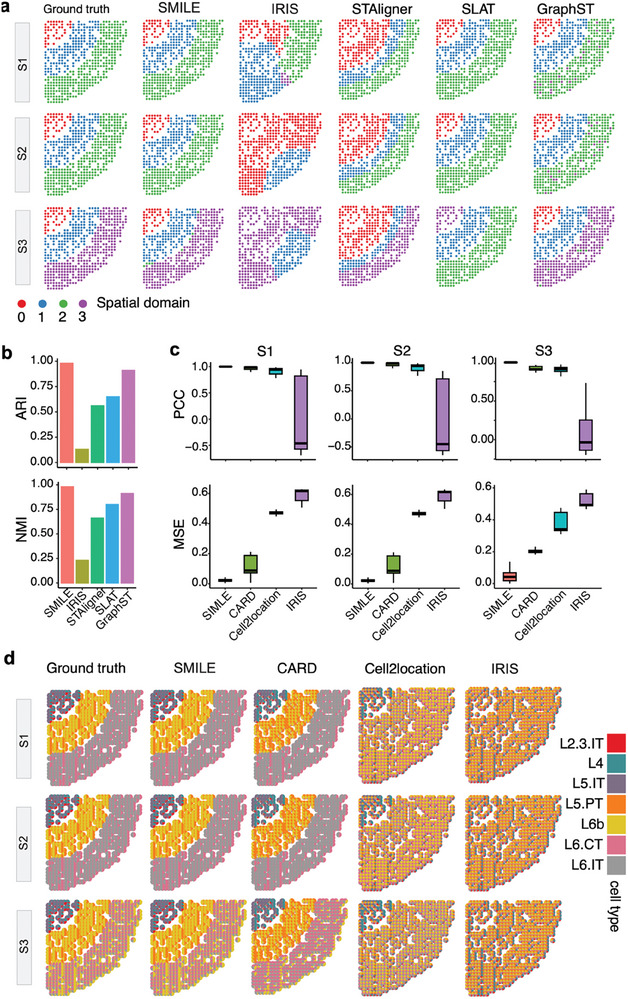
The spatial structure of cells is highly organized at multiscale levels from global spatial domains to local cell type heterogeneity. Existing methods for analyzing spatially resolved transcriptomics (SRT) are separately designed for either domain alignment across multiple slices or deconvoluting cell type compositions within a single slice. To this end, a novel deep learning method, SMILE, is proposed which combines graph contrastive autoencoder and multilayer perceptron with local constraints to learn multiscale and informative spot representations. By comparing SMILE with the state-of-the-art methods on simulation and real datasets, the superior performance of SMILE is demonstrated on spatial alignment, domain identification, and cell type deconvolution. The results show SMILE's capability not only in simultaneously dissecting spatial variations at different scales but also in unraveling altered cellular microenvironments in diseased conditions. Moreover, SMILE can utilize prior domain annotation information of one slice to further enhance the performance.
Keywords: cell type deconvolution; multiscale structure; scRNA‐seq; spatial domain; spatial transcriptomics.
© 2025 The Author(s). Advanced Science published by Wiley‐VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
-
- Ståhl P. L., Salmén F., Vickovic S., Lundmark A., Navarro J. F., Magnusson J., Giacomello S., Asp M., Westholm J. O., Huss M., Mollbrink A., Linnarsson S., Codeluppi S., Borg Å., Pontén F., Costea P. I., Sahlén P., Mulder J., Bergmann O., Lundeberg J., Frisén J., Science 2016, 353, 78. - PubMed
-
- Chen A., Liao S., Cheng M., Ma K., Wu L., Lai Y., Qiu X., Yang J., Xu J., Hao S., Wang X., Lu H., Chen X. i., Liu X., Huang X., Li Z., Hong Y., Jiang Y., Peng J., Liu S., Shen M., Liu C., Li Q., Yuan Y., Wei X., Zheng H., Feng W., Wang Z., Liu Y., Wang Z., et al., Cell 2022, 185, 1777. - PubMed
-
- Danishuddin S. K., Kim J. J., Drug Discov. Today 2024, 29, 103889. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources