

Cancer-independent somatic mutation of the wild-type NF1 allele in normal tissues in neurofibromatosis type 1
- PMID: 40000831
- PMCID: PMC11906363
- DOI: 10.1038/s41588-025-02097-2
Cancer-independent somatic mutation of the wild-type NF1 allele in normal tissues in neurofibromatosis type 1
Abstract
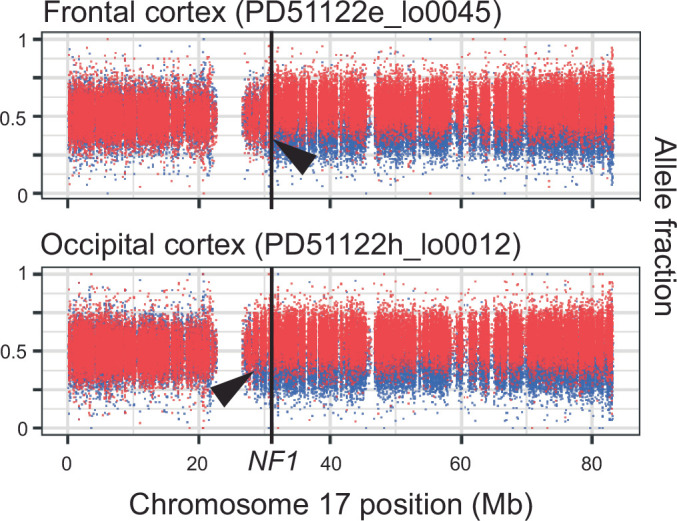
Cancer predisposition syndromes mediated by recessive cancer genes generate tumors via somatic variants (second hits) in the unaffected allele. Second hits may or may not be sufficient for neoplastic transformation. Here we performed whole-genome and whole-exome sequencing on 479 tissue biopsies from a child with neurofibromatosis type 1, a multisystem cancer-predisposing syndrome mediated by constitutive monoallelic NF1 inactivation. We identified multiple independent NF1 driver variants in histologically normal tissues, but not in 610 biopsies from two nonpredisposed children. We corroborated this finding using targeted duplex sequencing, including a further nine adults with the same syndrome. Overall, truncating NF1 mutations were under positive selection in normal tissues from individuals with neurofibromatosis type 1. We demonstrate that normal tissues in neurofibromatosis type 1 commonly harbor second hits in NF1, the extent and pattern of which may underpin the syndrome's cancer phenotype.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: I.M. is a cofounder and consultant of Quotient Therapeutics. D.H. provides consultancy to AstraZeneca/MedImmune, Alexion Pharmaceuticals, Bayer, Biodexa, Roche/Genentech and Novartis, as well as expert testimony to AstraZeneca and Novartis, and his expenses are covered by Alexion Pharmaceuticals, Boehringer Ingelheim, Roche/Genentech and Novartis. The other authors declare no competing interests.
Figures








References
-
- Lee-Six, H. et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature574, 532–537 (2019). - PubMed
-
- Moore, L. et al. The mutational landscape of human somatic and germline cells. Nature597, 381–386 (2021). - PubMed
-
- Lawson, A. R. J. et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science370, 75–82 (2020). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
