

Digital sequencing is improved by using structured unique molecular identifiers
- PMID: 40001095
- PMCID: PMC11853513
- DOI: 10.1186/s13059-025-03504-x
Digital sequencing is improved by using structured unique molecular identifiers
Abstract
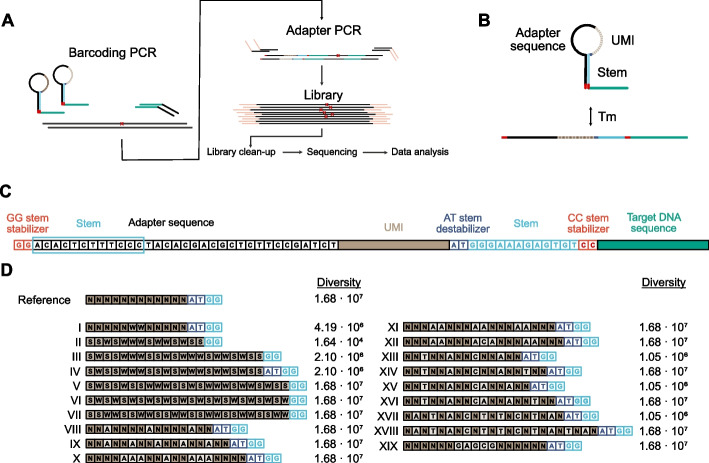
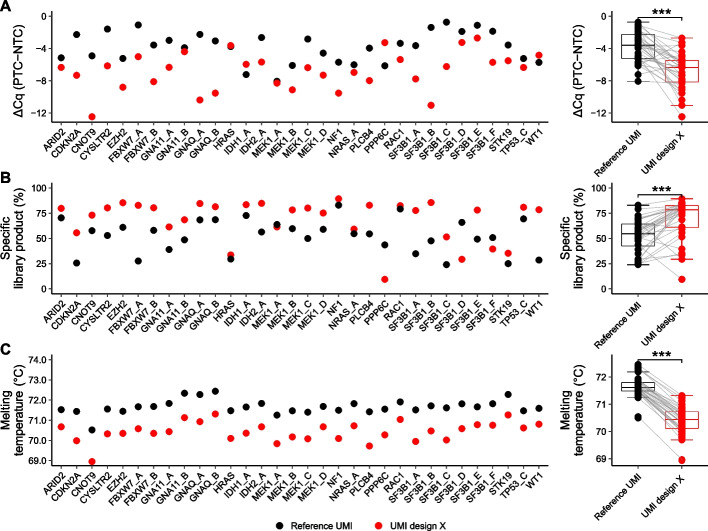
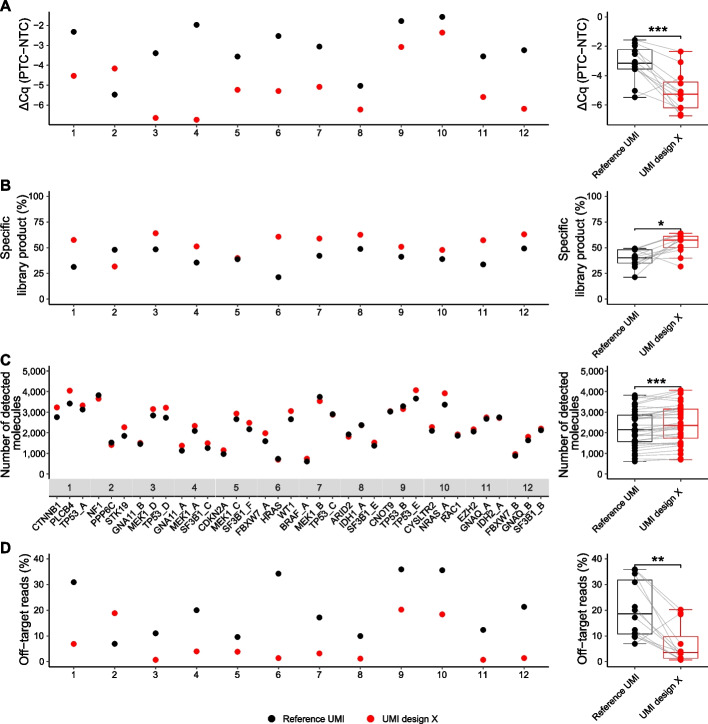
Digital sequencing uses unique molecular identifiers (UMIs) to correct for polymerase induced errors and amplification biases. Here, we design 19 different structured UMIs to minimize the capacity of primers to form non-specific PCR products during library construction using SiMSen-Seq, a PCR-based digital sequencing approach with flexible multiplexing capabilities suitable for tumor-informed mutation analysis. All structured UMI designs demonstrate enhanced assay performance compared with an unstructured reference UMI. The best performing structured UMI design shows significant improvements in all tested aspects of assay and sequencing performance with the ability to reliable detect low variant allele frequencies.
Keywords: Digital sequencing; Error-free sequencing; Molecular barcode; Sequencing; Unique molecular identifier.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: This study was performed in accordance with the Declaration of Helsinki and approved by the Regional Ethical Review Board in Gothenburg (DNR 485–16, approved 23 June 2016; DNR T-795–16, approved 15 September 2016; DNR T525-18, approved 5 June 2018; and DNR 2021–04895, approved 24 October 2021). Informed consent was obtained. Consent for publication: The study subject has consented for publication. Competing interests: The use of structured unique molecular identifiers in SiMSen-Seq is patent pending. S.F. is employed by SiMSen Diagnostics. G.J. declares employment and stock ownership in SiMSen Diagnostics. A.S. is co-inventor of the SiMSen-Seq technology that is patent protected (U.S. Serial No.: 15/552,618). A.S. declares stock ownership in Tulebovaasta, Iscaff Pharma, and SiMSen Diagnostics and is a board member in Tulebovaasta.
Figures



References
MeSH terms
LinkOut - more resources
Full Text Sources
