

Iron Accumulation and Lipid Peroxidation in Cellular Models of Nemaline Myopathies
- PMID: 40003902
- PMCID: PMC11855326
- DOI: 10.3390/ijms26041434
Iron Accumulation and Lipid Peroxidation in Cellular Models of Nemaline Myopathies
Abstract
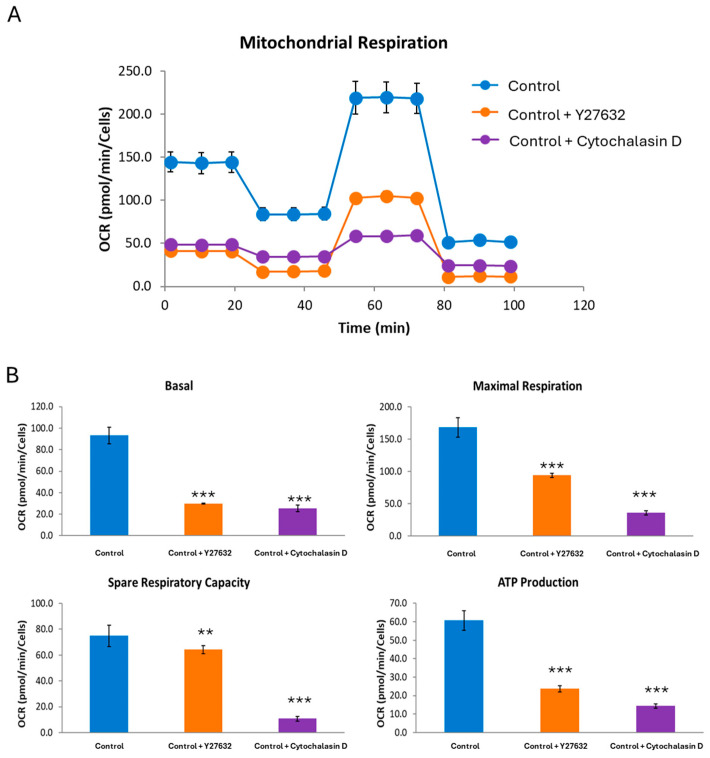
One of the most prevalent types of congenital myopathy is nemaline myopathy (NM), which is recognized by histopathological examination of muscle fibers for the presence of "nemaline bodies" (rods). Mutations in the actin alpha 1 (ACTA1) and nebulin (NEB) genes result in the most prevalent types of NM. Muscle weakness and hypotonia are the main clinical characteristics of this disease. Unfortunately, the pathogenetic mechanisms are still unknown, and there is no cure. In previous work, we showed that actin filament polymerization defects in patient-derived fibroblasts were associated with mitochondrial dysfunction. In this manuscript, we examined the pathophysiological consequences of mitochondrial dysfunction in patient-derived fibroblasts. We analyzed iron and lipofuscin accumulation and lipid peroxidation both at the cellular and mitochondrial level. We found that fibroblasts derived from patients harboring ACTA1 and NEB mutations showed intracellular iron and lipofuscin accumulation, increased lipid peroxidation, and altered expression levels of proteins involved in iron metabolism. Furthermore, we showed that actin polymerization inhibition in control cells recapitulates the main pathological alterations of mutant nemaline cells. Our results indicate that mitochondrial dysfunction is associated with iron metabolism dysregulation, leading to iron/lipofuscin accumulation and increased lipid peroxidation.
Keywords: iron accumulation; lipid peroxidation; nemaline myopathy.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures














References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
