

Analysis of Short Tandem Repeat Expansions in a Cohort of 12,496 Exomes from Patients with Neurological Diseases Reveals Variable Genotyping Rate Dependent on Exome Capture Kits
- PMID: 40004498
- PMCID: PMC11855749
- DOI: 10.3390/genes16020169
Analysis of Short Tandem Repeat Expansions in a Cohort of 12,496 Exomes from Patients with Neurological Diseases Reveals Variable Genotyping Rate Dependent on Exome Capture Kits
Abstract
Background/objectives: Short tandem repeat expansions are the most common cause of inherited neurological diseases. These disorders are clinically and genetically heterogeneous, such as in myotonic dystrophy and spinocerebellar ataxia, and they are caused by different repeat motifs in different genomic locations. Major advances in bioinformatic tools used to detect repeat expansions from short read sequencing data in the last few years have led to the implementation of these workflows into next generation sequencing pipelines in healthcare. Here, we aimed to evaluate the clinical utility of analysing repeat expansions through exome sequencing in a large cohort of genetically undiagnosed patients with neurological disorders.
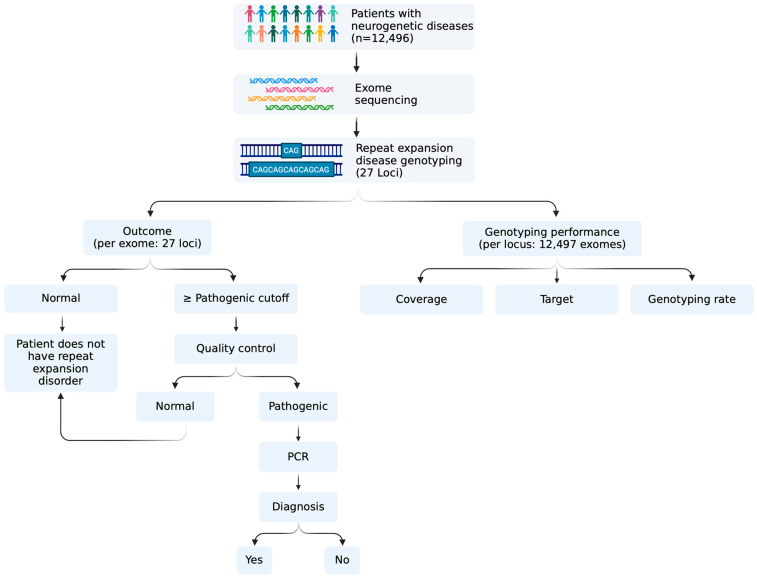
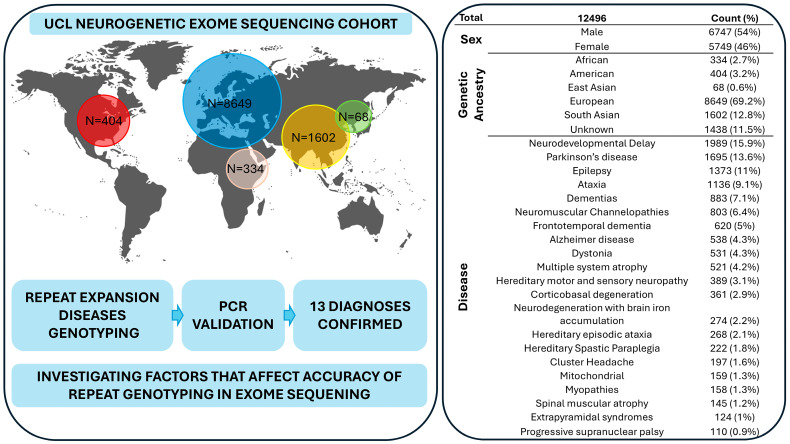
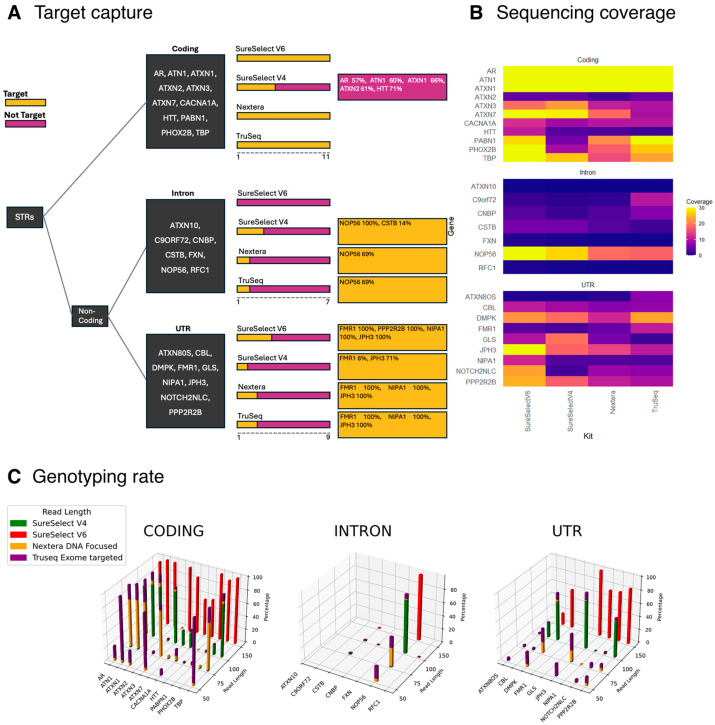
Methods: We here analyse 27 disease-causing DNA repeats found in the coding, intronic and untranslated regions in 12,496 exomes in patients with a range of neurogenetic conditions.
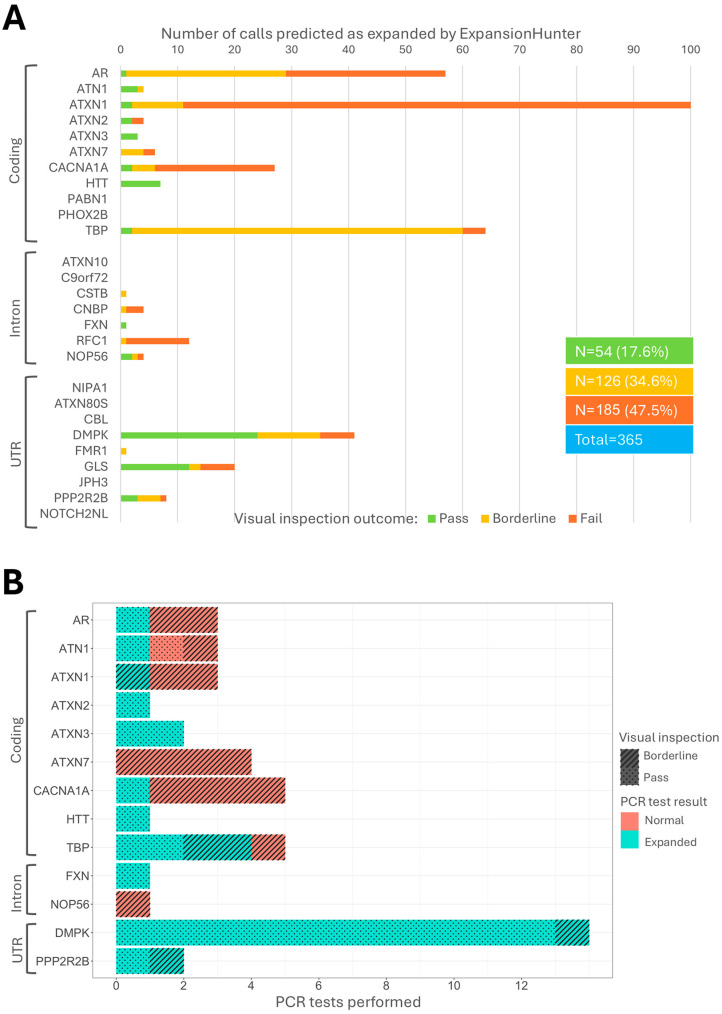
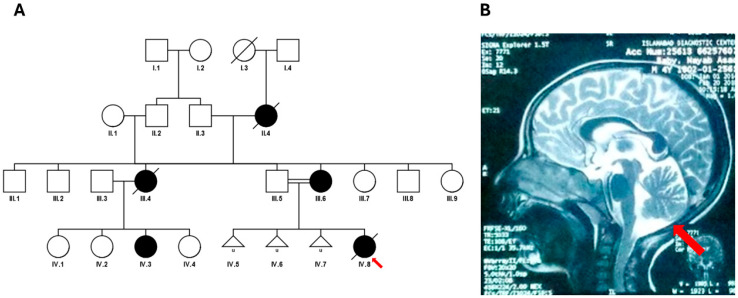
Results: We identified-and validated by polymerase chain reaction-29 repeat expansions across a range of loci, 48% (n = 14) of which were diagnostic. We then analysed the genotyping performance across all repeat loci and found that, despite high coverage in most repeats in coding regions, some loci had low genotyping rates, such as those that cause spinocerebellar ataxia 2 (ATXN2, 0.1-8.4%) and Huntington disease (HTT, 0.2-58.2%), depending on the capture kit. Conversely, while most intronic repeats were not genotyped, we found a high genotyping rate in the intronic locus that causes spinocerebellar ataxia 36 (NOP56, 30.1-98.3%) and in the one that causes myotonic dystrophy type 1 (DMPK, myotonic dystrophy type 1).
Conclusions: We show that the key factors that influence the genotyping rate of repeat expansion loci analysis are the sequencing read length and exome capture kit. These results provide important information about the performance of exome sequencing as a genetic test for repeat expansion disorders.
Keywords: ExpansionHunter; Huntington disease; exome sequencing; myotonic dystrophy; repeat expansion disease; repeat expansion diseases; short read sequencing; short tandem repeat; spinocerebellar ataxia.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Ibañez K., Polke J., Hagelstrom R.T., Dolzhenko E., Pasko D., Thomas E.R.A., Daugherty C.L., Kasperaviciute D., Smith R.K., WGS for Neurological Diseases Group et al. Whole genome sequencing for the diagnosis of neurological repeat expansion disorders in the UK: A retrospective diagnostic accuracy and prospective clinical validation study. Lancet Neurol. 2022;21:234–245. doi: 10.1016/S1474-4422(21)00462-2. - DOI - PMC - PubMed
-
- Van der Sanden B.P.G.H., Corominas J., De Groot M., Pennings M., Meijer R.P.P., Verbeek N., Van de Warrenburg B., Schouten M., Yntema G.H., Vissers E.L.M.L., et al. Systematic analysis of short tandem repeats in 38,095 exomes provides an additional diagnostic yield. Genet. Med. 2021;23:1569–1573. doi: 10.1038/s41436-021-01174-1. - DOI - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
