

Hereditary, non HINT1 related, axonal neuropathy with neuromyotonia
- PMID: 40009145
- PMCID: PMC12084164
- DOI: 10.1007/s10072-025-08022-z
Hereditary, non HINT1 related, axonal neuropathy with neuromyotonia
Abstract
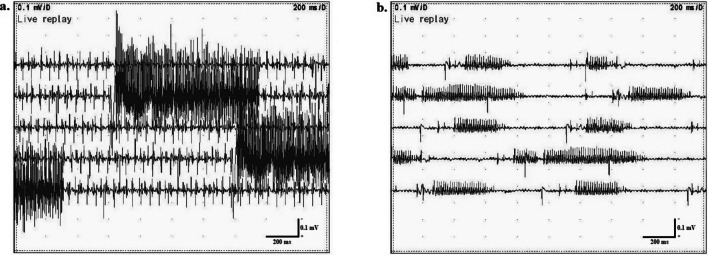
To date, neuromyotonia in the context of an inherited axonal neuropathy has been linked to autosomal recessive mutations in the histidine triad nucleotide binding protein 1 (HINT1) gene. In this study we describe two unrelated male patients with late-onset, predominantly motor, axonal neuropathy with neuromyotonia, who carried an autosomal dominant c.103G > A mutation in the myelin protein zero (MPZ) gene (NM_000530.8:c.103G > A, p.Asp35Asn), identified by whole-exome sequence analysis (WES). CASE DESCRIPTIONS: The first patient presented progressive leg muscle weakness and stiffness with difficulty in walking, pain and increased creatine kinase levels,during his fifth decade of life. Electrophysiological examination revealed findings of an axonal, length-dependent polyneuropathy with spontaneous activity, mainly neuromyotonia. Over the 20-year disease course since the first reported symptoms, muscle weakness gradually worsened and he is currently unable to walk without assistance. A second male patient, unrelated to the first one, showed similar clinical and electrophysiological features of a length-dependent axonal neuropathy with neuromyotonia. WES detected the same MPZ missensevariant. CONCLUSION: This study suggests a novel entity in the spectrum of Charcot-Marie-Tooth hereditary neuropathies, characterized by autosomal dominant axonal neuropathy with neuromyotonia (AD-NMAN).
Keywords: Axonal neuropathy; Charcot-Marie-tooth; Myelin protein zero; Neuromuscular ultrasound; Neuromyotonia.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Informed consent: Informed consent was obtained from the patients. Competing interests: None of the authors have potential conflicts of interest to be disclosed. Ethical approval: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Study approval was obtained by the Research and Ethics Committee of the University Hospital of Patras (no. of approval 8482/15.03.2024).
Figures
References
-
- Bashford J, Chan WK, Coutinho E, Norwood F, Mills K, Shaw CE (2021) Demystifying the spontaneous phenomena of motor hyperexcitability. Clin Neurophysiol 132(8):1830–1844. 10.1016/j.clinph.2021.03.053 - PubMed
-
- Carroll AS, Burns J, Nicholson G, Kiernan MC, Vucic S, Neuropathies I (2019) Semin Neurol 39(5):620–639. 10.1055/s-0039-1693006 - PubMed
-
- Callegari I, Gemelli C, Geroldi A, Veneri F, Mandich P, D’Antonio M, Pareyson D, Shy ME, Schenone A, Prada V, Grandis M (2019) Mutation update for myelin protein zero-related neuropathies and the increasing role of variants causing a late-onset phenotype. J Neurol 266(11):2629–2645. 10.1007/s00415-019-09453-3 - PubMed
-
- Fridman V, Sillau S, Bockhorst J, Smith K, Moroni I, Pagliano E, Pisciotta C, Piscosquito G, Laurá M, Muntoni F, Bacon C, Feely S, Grider T, Gutmann L, Shy R, Wilcox J, Herrmann DN, Li J, Ramchandren S, Sumner CJ, Lloyd TE, Day J, Siskind CE, Yum SW, Sadjadi R, Finkel RS, Scherer SS, Pareyson D, Reilly MM, Shy ME (2023) Disease Progression in Charcot-Marie-tooth disease related to MPZ mutations: a longitudinal study. Ann Neurol 93(3):563–576. 10.1002/ana.26518 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
