

Decoding heterogeneous single-cell perturbation responses
- PMID: 40011559
- PMCID: PMC11906366
- DOI: 10.1038/s41556-025-01626-9
Decoding heterogeneous single-cell perturbation responses
Abstract
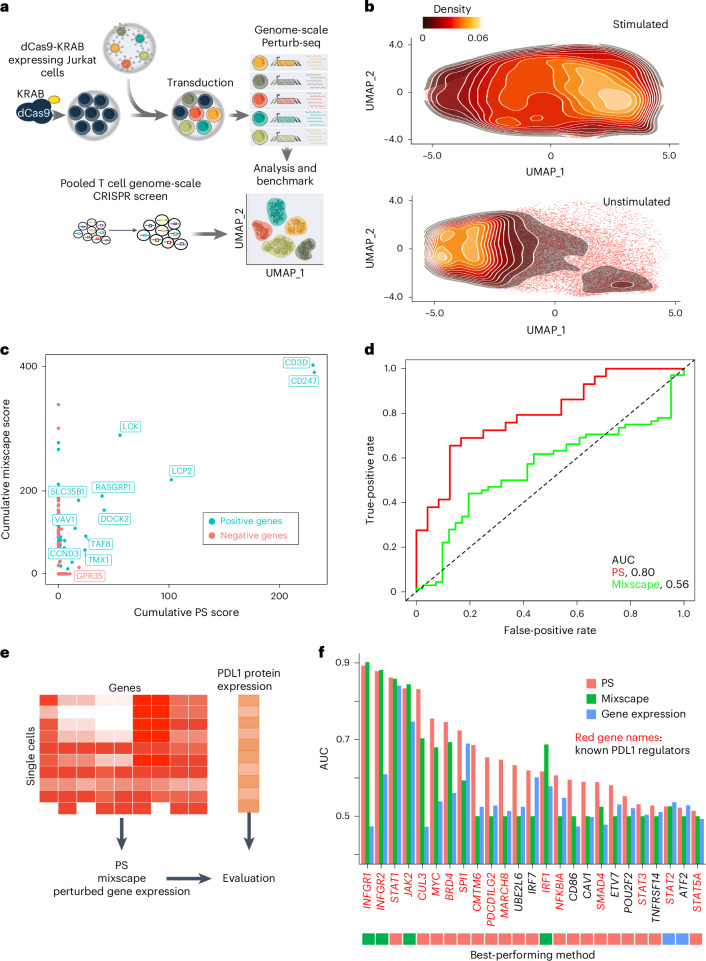
Understanding how cells respond differently to perturbation is crucial in cell biology, but existing methods often fail to accurately quantify and interpret heterogeneous single-cell responses. Here we introduce the perturbation-response score (PS), a method to quantify diverse perturbation responses at a single-cell level. Applied to single-cell perturbation datasets such as Perturb-seq, PS outperforms existing methods in quantifying partial gene perturbations. PS further enables single-cell dosage analysis without needing to titrate perturbations, and identifies 'buffered' and 'sensitive' response patterns of essential genes, depending on whether their moderate perturbations lead to strong downstream effects. PS reveals differential cellular responses on perturbing key genes in contexts such as T cell stimulation, latent HIV-1 expression and pancreatic differentiation. Notably, we identified a previously unknown role for the coiled-coil domain containing 6 (CCDC6) in regulating liver and pancreatic cell fate decisions. PS provides a powerful method for dose-to-function analysis, offering deeper insights from single-cell perturbation data.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: T.B. is a cofounder and Managing Director of Myllia Biotechnology. A.K., A.V., N.U. and A.L. are employees of Myllia Biotechnology. The other authors declare no competing interests.
Figures















Update of
-
Decoding Heterogenous Single-cell Perturbation Responses.bioRxiv [Preprint]. 2023 Nov 29:2023.10.30.564796. doi: 10.1101/2023.10.30.564796. bioRxiv. 2023. Update in: Nat Cell Biol. 2025 Mar;27(3):493-504. doi: 10.1038/s41556-025-01626-9. PMID: 37961332 Free PMC article. Updated. Preprint.
References
-
- Jaitin, D. A. et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell167, 1883–1896.e15 (2016). - PubMed
MeSH terms
Grants and funding
- R35 GM140888/GM/NIGMS NIH HHS/United States
- UM1 HG012654/HG/NHGRI NIH HHS/United States
- DBI-1846216, DMS-2113754/National Science Foundation (NSF)
- R01HL168174/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01HG010753/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
