

Programs, origins and immunomodulatory functions of myeloid cells in glioma
- PMID: 40011771
- PMCID: PMC12018266
- DOI: 10.1038/s41586-025-08633-8
Programs, origins and immunomodulatory functions of myeloid cells in glioma
Abstract
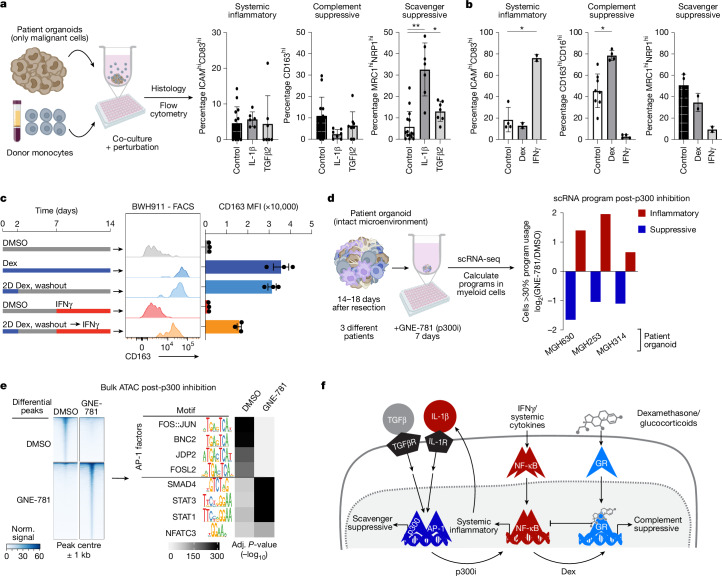
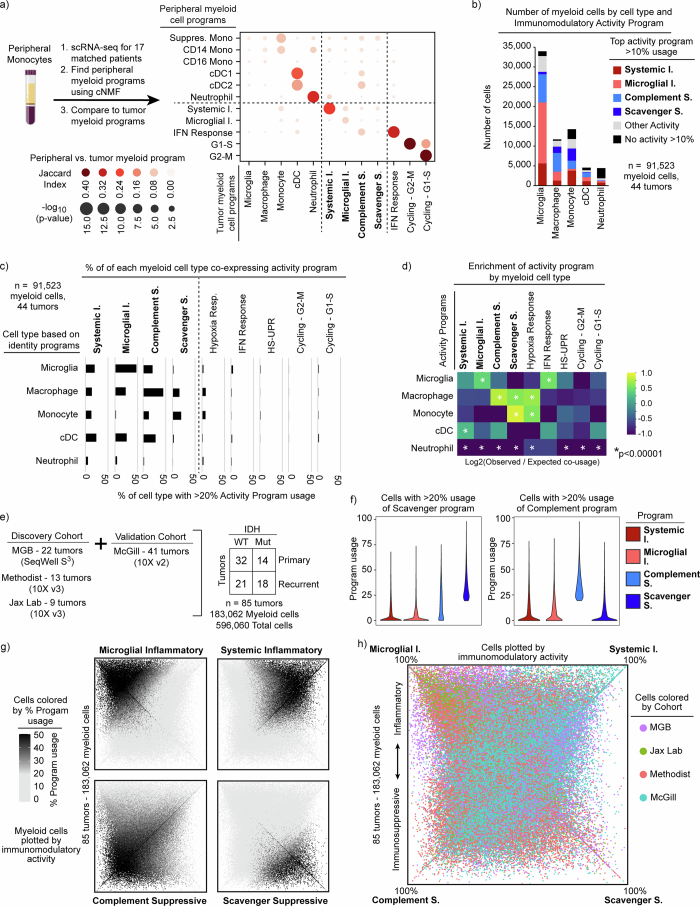
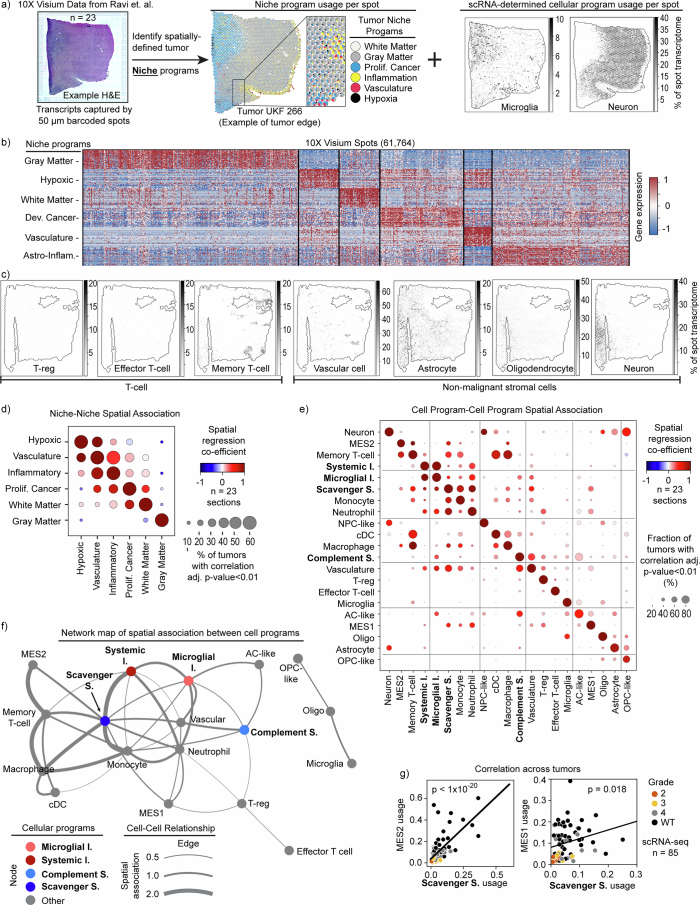
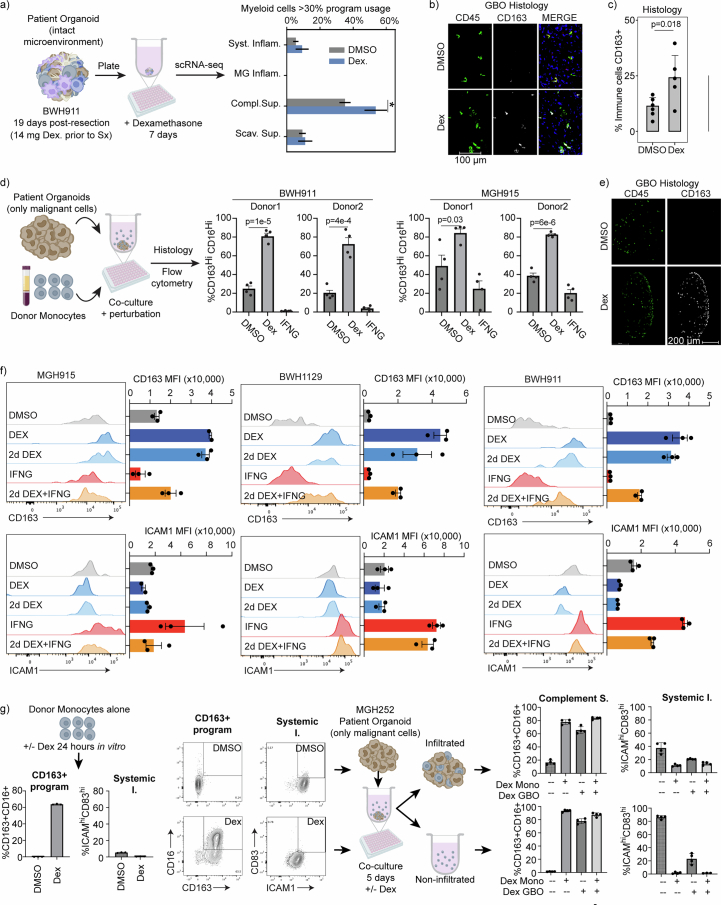
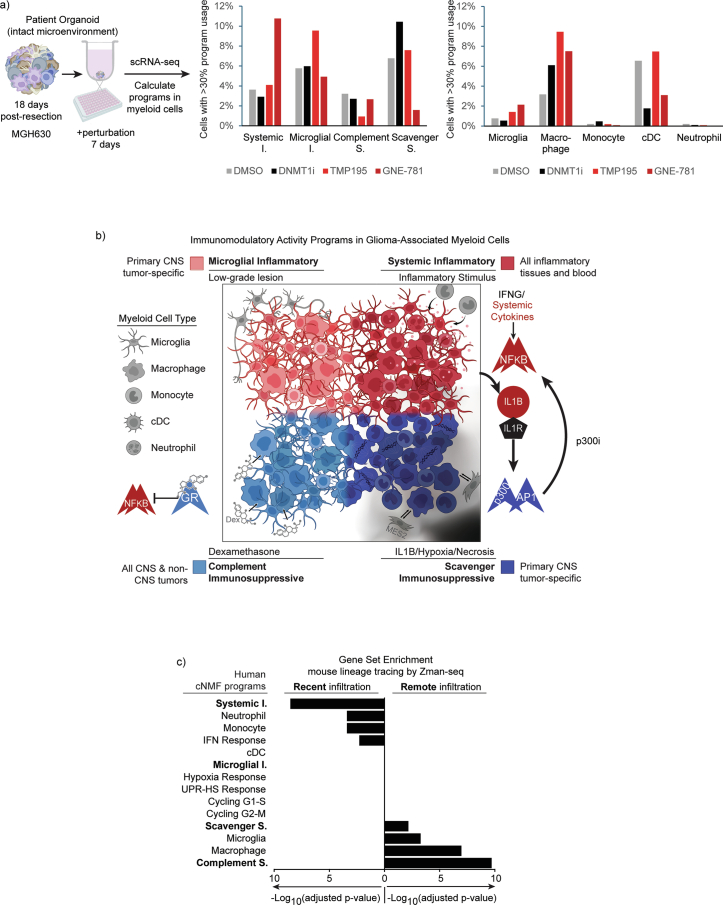
Gliomas are incurable malignancies notable for having an immunosuppressive microenvironment with abundant myeloid cells, the immunomodulatory phenotypes of which remain poorly defined1. Here we systematically investigate these phenotypes by integrating single-cell RNA sequencing, chromatin accessibility, spatial transcriptomics and glioma organoid explant systems. We discovered four immunomodulatory expression programs: microglial inflammatory and scavenger immunosuppressive programs, which are both unique to primary brain tumours, and systemic inflammatory and complement immunosuppressive programs, which are also expressed by non-brain tumours. The programs are not contingent on myeloid cell type, developmental origin or tumour mutational state, but instead are driven by microenvironmental cues, including tumour hypoxia, interleukin-1β, TGFβ and standard-of-care dexamethasone treatment. Their relative expression can predict immunotherapy response and overall survival. By associating the respective programs with mediating genomic elements, transcription factors and signalling pathways, we uncover strategies for manipulating myeloid-cell phenotypes. Our study provides a framework to understand immunomodulation by myeloid cells in glioma and a foundation for the development of more-effective immunotherapies.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: T.E.M. has financial interests in Reify Health, Care Access Research and Telomere Diagnostics. C.P.C. has financial interest in Axoft. L.N.G.C. receives consulting fees from Elsevier, Oakstone Publishing, Prime Education, BMJ Best Practice and Servier, and research funding from Merck & Co (to the Dana-Farber Cancer Institute). J.L.G. is consultant and serves on the scientific advisory board of Array BioPharma, AstraZeneca, BD Biosciences, Carisma, Codagenix, Duke Street Bio, GlaxoSmithKline, Kowa, Kymera, OncoOne and Verseau Therapeutics, and receives research support from Array BioPharma/Pfizer, Eli Lilly, GlaxoSmithKline and Merck. M.L.S. is an equity holder, scientific co-founder and advisory board member of Immunitas Therapeutics. A.K.S. receives compensation for consulting and/or scientific advisory board membership from Honeycomb Biotechnologies, Cellarity, Ochre Bio, Relation Therapeutics, FL86/Quotient Therapeutics, Parabalis Medicines, Passkey Therapeutics, IntrECate Biotherapeutics, Danaher, Senda Biosciences and Dahlia Biosciences that is unrelated to this work. B.E.B. has financial interests in Fulcrum Therapeutics, HiFiBio, Arsenal Biosciences, Chroma Medicine and Cell Signaling Technologies. The other authors declare no competing interests.
Figures












Update of
-
Programs, Origins, and Niches of Immunomodulatory Myeloid Cells in Gliomas.bioRxiv [Preprint]. 2023 Oct 27:2023.10.24.563466. doi: 10.1101/2023.10.24.563466. bioRxiv. 2023. Update in: Nature. 2025 Apr;640(8060):1072-1082. doi: 10.1038/s41586-025-08633-8. PMID: 37961527 Free PMC article. Updated. Preprint.
References
-
- Sørensen, M. D., Dahlrot, R. H., Boldt, H. B., Hansen, S. & Kristensen, B. W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol.44, 185–206 (2018). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
