

Genome-coverage single-cell histone modifications for embryo lineage tracing
- PMID: 40011786
- PMCID: PMC12003199
- DOI: 10.1038/s41586-025-08656-1
Genome-coverage single-cell histone modifications for embryo lineage tracing
Abstract
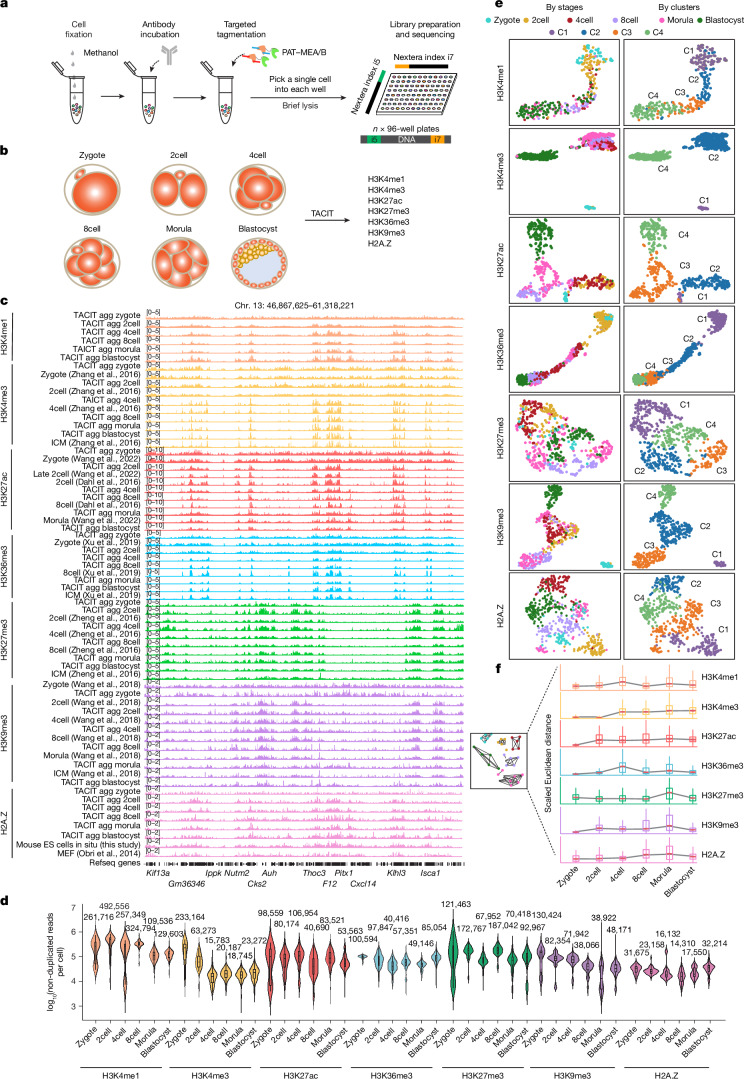
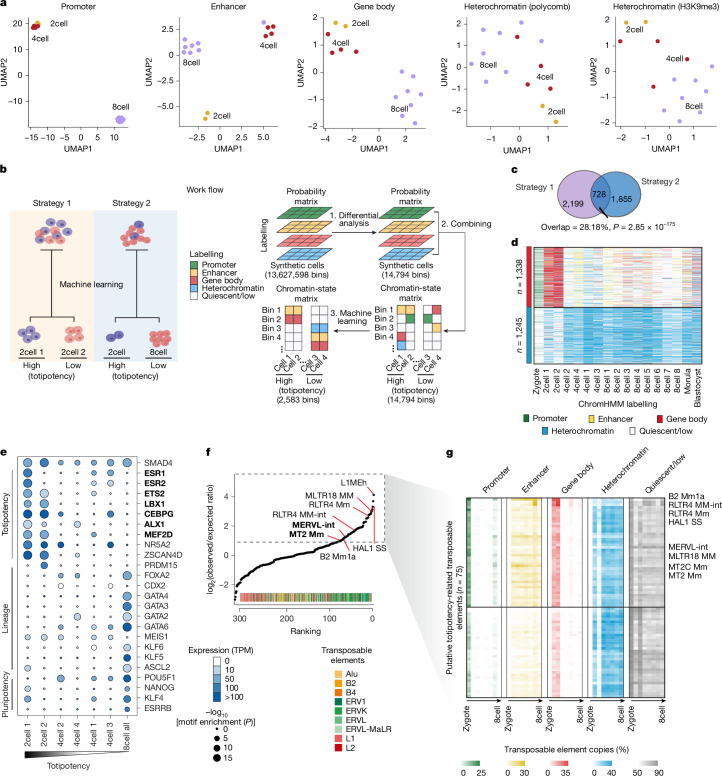
Substantial epigenetic resetting during early embryo development from fertilization to blastocyst formation ensures zygotic genome activation and leads to progressive cellular heterogeneities1-3. Mapping single-cell epigenomic profiles of core histone modifications that cover each individual cell is a fundamental goal in developmental biology. Here we develop target chromatin indexing and tagmentation (TACIT), a method that enabled genome-coverage single-cell profiling of seven histone modifications across mouse early embryos. We integrated these single-cell histone modifications with single-cell RNA sequencing data to chart a single-cell resolution epigenetic landscape. Multimodal chromatin-state annotations showed that the onset of zygotic genome activation at the early two-cell stage already primes heterogeneities in totipotency. We used machine learning to identify totipotency gene regulatory networks, including stage-specific transposable elements and putative transcription factors. CRISPR activation of a combination of these identified transcription factors induced totipotency activation in mouse embryonic stem cells. Together with single-cell co-profiles of multiple histone modifications, we developed a model that predicts the earliest cell branching towards the inner cell mass and the trophectoderm in latent multimodal space and identifies regulatory elements and previously unknown lineage-specifying transcription factors. Our work provides insights into single-cell epigenetic reprogramming, multimodal regulation of cellular lineages and cell-fate priming during mouse pre-implantation development.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures













References
-
- Burton, A. & Torres-Padilla, M. E. Chromatin dynamics in the regulation of cell fate allocation during early embryogenesis. Nat. Rev. Mol. Cell Biol.15, 723–734 (2014). - PubMed
-
- Zhang, Y. & Xie, W. Building the genome architecture during the maternal to zygotic transition. Curr. Opin. Genet. Dev.72, 91–100 (2022). - PubMed
-
- Hemberger, M., Dean, W. & Reik, W. Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nat. Rev. Mol. Cell Biol.10, 526–537 (2009). - PubMed
-
- Zernicka-Goetz, M., Morris, S. A. & Bruce, A. W. Making a firm decision: multifaceted regulation of cell fate in the early mouse embryo. Nat. Rev. Genet.10, 467–477 (2009). - PubMed
-
- Posfai, E. et al. Evaluating totipotency using criteria of increasing stringency. Nat. Cell Biol.23, 49–60 (2021). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
