

A compendium of human gene functions derived from evolutionary modelling
- PMID: 40011791
- PMCID: PMC11964926
- DOI: 10.1038/s41586-025-08592-0
A compendium of human gene functions derived from evolutionary modelling
Abstract
A comprehensive, computable representation of the functional repertoire of all macromolecules encoded within the human genome is a foundational resource for biology and biomedical research. The Gene Ontology Consortium has been working towards this goal by generating a structured body of information about gene functions, which now includes experimental findings reported in more than 175,000 publications for human genes and genes in experimentally tractable model organisms1,2. Here, we describe the results of a large, international effort to integrate all of these findings to create a representation of human gene functions that is as complete and accurate as possible. Specifically, we apply an expert-curated, explicit evolutionary modelling approach to all human protein-coding genes. This approach integrates available experimental information across families of related genes into models that reconstruct the gain and loss of functional characteristics over evolutionary time. The models and the resulting set of 68,667 integrated gene functions cover approximately 82% of human protein-coding genes. The functional repertoire reveals a marked preponderance of molecular regulatory functions, and the models provide insights into the evolutionary origins of human gene functions. We show that our set of descriptions of functions can improve the widely used genomic technique of Gene Ontology enrichment analysis. The experimental evidence for each functional characteristic is recorded, thereby enabling the scientific community to help review and improve the resource, which we have made publicly available.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures
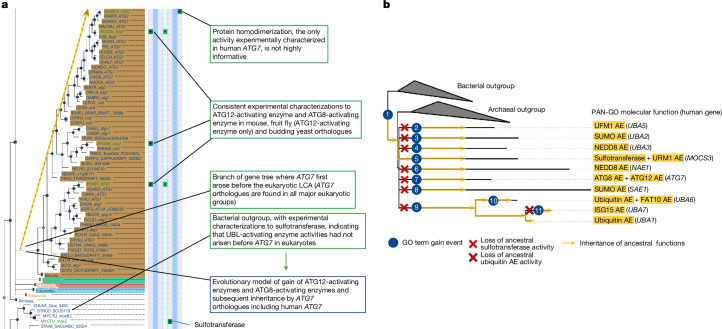


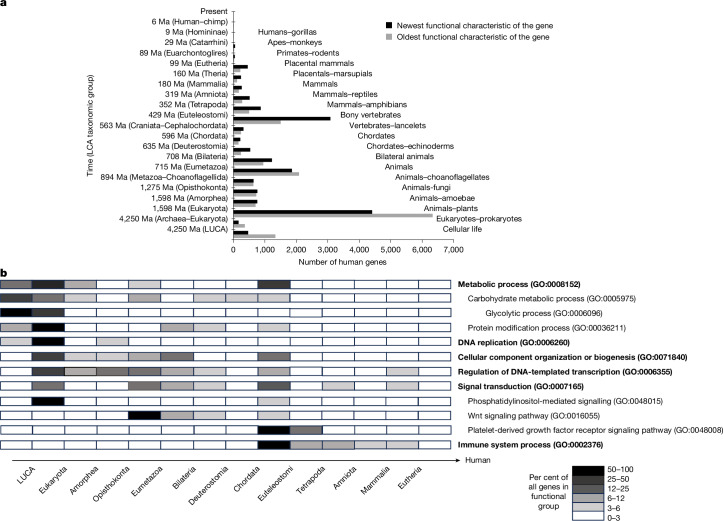



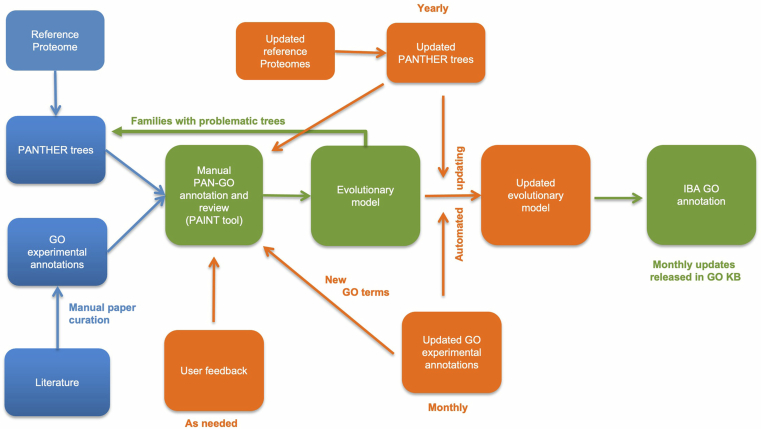

References
-
- Fields, S. & Johnston, M. Cell biology. Whither model organism research? Science307, 1885–1886 (2005). - PubMed
-
- Müller, B. & Grossniklaus, U. Model organisms—a historical perspective. J. Proteomics73, 2054–2063 (2010). - PubMed
-
- Venter, J. C. et al. The sequence of the human genome. Science291, 1304–1351 (2001). - PubMed
-
- Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature409, 860–921 (2001). - PubMed
MeSH terms
Grants and funding
- U24 HG000330/HG/NHGRI NIH HHS/United States
- U24 HG002659/HG/NHGRI NIH HHS/United States
- U41 HG000739/HG/NHGRI NIH HHS/United States
- U24 HG010859/HG/NHGRI NIH HHS/United States
- U24 HG012212/HG/NHGRI NIH HHS/United States
- U24 HG007822/HG/NHGRI NIH HHS/United States
- U24 HG001315/HG/NHGRI NIH HHS/United States
- P41 HG002273/HG/NHGRI NIH HHS/United States
- U24 HG002223/HG/NHGRI NIH HHS/United States
- R01 HL064541/HL/NHLBI NIH HHS/United States
- U41 HG002273/HG/NHGRI NIH HHS/United States
- P41 HD064556/HD/NICHD NIH HHS/United States
- R01 HG002273/HG/NHGRI NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
