

CACNA1S-associated triadopathy presenting with myalgia, muscle weakness, and asymptomatic hyperCKemia
- PMID: 40018084
- PMCID: PMC11866399
- DOI: 10.1177/17562864251317961
CACNA1S-associated triadopathy presenting with myalgia, muscle weakness, and asymptomatic hyperCKemia
Abstract
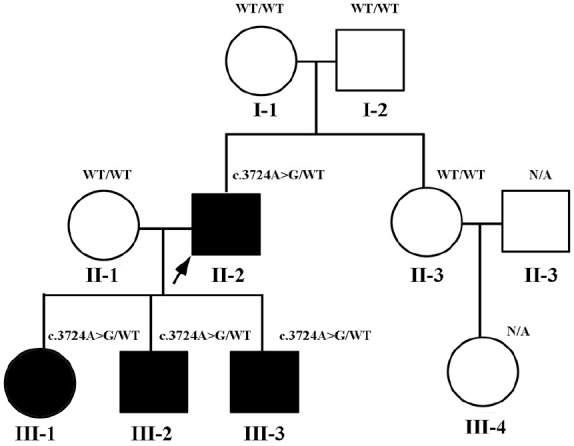
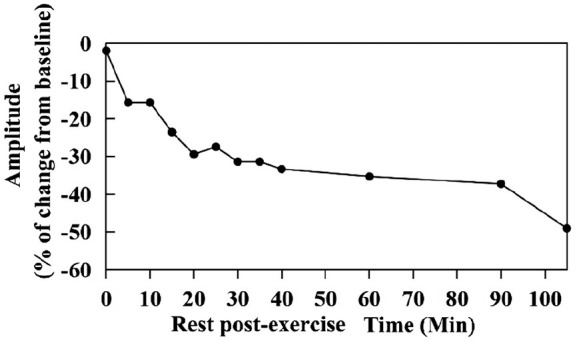
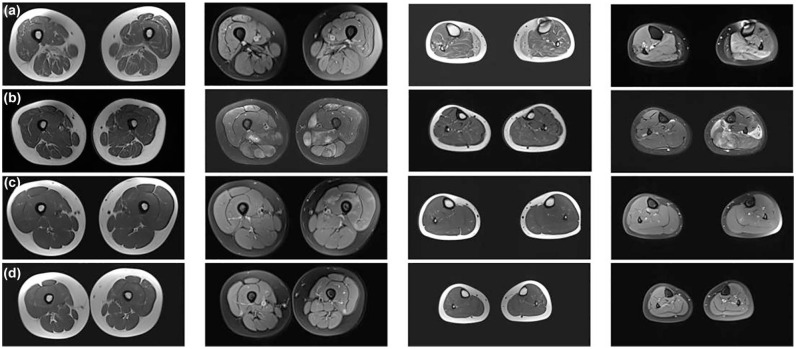
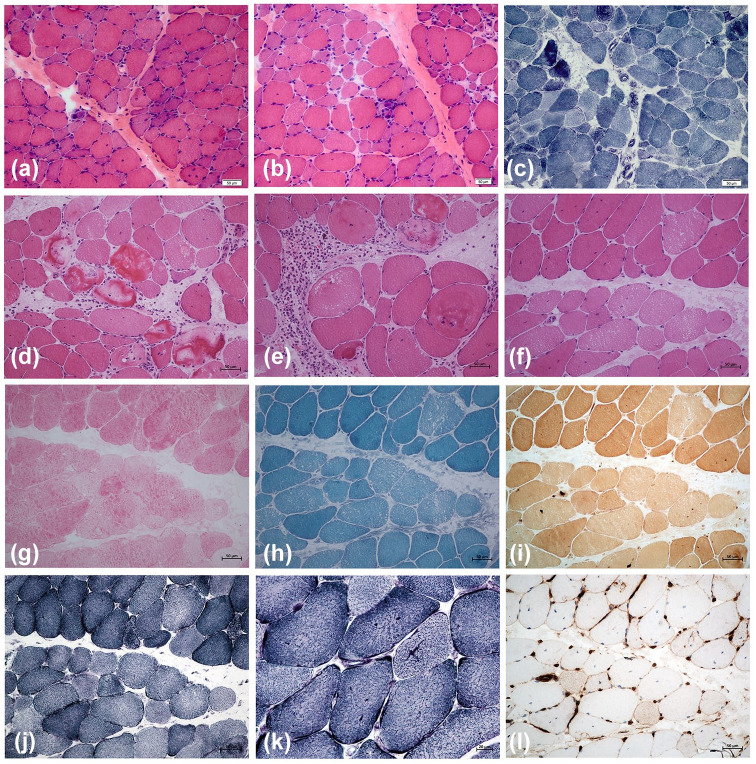
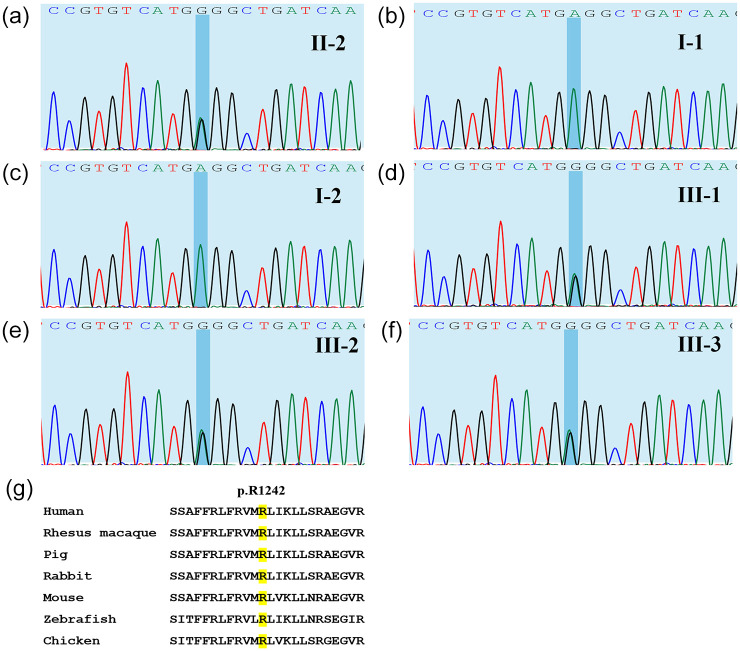
CACNA1S variants can alter the structure and function of the calcium channel, resulting in abnormal calcium influx and homeostasis. It is well established that pathogenic variants in CACNA1S can lead to hypokalemic periodic paralysis, malignant hyperthermia, and congenital myopathy. Nevertheless, the clinical presentations and disease progression of exertional myalgia and weakness associated with CACNA1S variants remain elusive. In this study, four affected individuals from an autosomal-dominant family were described, exhibiting symptoms of severe exertional myalgia, followed by flaccid weakness or rhabdomyolysis, along with asymptomatic hyperCKemia during the interictal period. Long exercise test showed a late decrease in compound muscle action potential amplitude. Muscle MRI revealed edema-like changes in the early stage, and fatty degeneration and substitution in prolonged disease courses, while closely aligned with the features of chronic myopathy. Ultrastructural examination revealed dilation of the sarcoplasmic reticulum and myofibrillar structural disarrangement. Genetic screening identified a c.3724A>G (p.Arg1242Gly) mutation in the CACNA1S gene. A literature review revealed that 15 patients exhibited the exertional myalgia and weakness phenotype associated with CACNA1S mutations, presenting similar clinical, electrophysiological, radiological, and pathological features. As the disease progressed, these patients developed severe muscle weakness, ultimately leading to wheelchair dependency. This exertional myalgia-weakness phenotype represented a unique CACNA1S-related phenotype that broadened the spectrum of CACNA1S-associated myopathy, bridging between periodic paralysis and congenital myopathies. The similarities between CACNA1S-associated myalgia-weakness and RyR1-associated myalgia-weakness underscored a shared pathogenesis of excitatory-contractile coupling at the triad of skeletal muscle.
Keywords: CACNA1S; hyperCKemia; myalgia; ryanodine receptor; triad.
Plain language summary
CACNA1S-linked muscle disorder with pain, weakness, and asymptomatic high creatine kinase levels The CACNA1S gene is linked to several muscle conditions, including hypokalemic periodic paralysis (HypoPP), malignant hyperthermia, and congenital myopathy. In this study, four people from a family with an inherited pattern of the disease were examined. They experienced severe muscle pain after exercise, followed by muscle weakness or muscle breakdown (rhabdomyolysis), and had elevated levels of a protein called CK in their blood when they were not having symptoms. Genetic testing found a mutation (change) in the CACNA1S gene: c.3724A>G (p.Arg1242Gly). A review of other cases showed that 15 people with similar symptoms—exercise-related muscle pain and weakness—also had mutations in the CACNA1S gene. These patients showed similar signs in tests of their muscle function, imaging scans, and tissue samples. This pattern of muscle pain and weakness is a new type of CACNA1S-related muscle disease, expanding our understanding of how mutations in this gene can cause different kinds of muscle problems. It helps link conditions like periodic paralysis and congenital myopathies.
© The Author(s), 2025.
Figures

Similar articles
-
Rhabdomyolysis and fluctuating asymptomatic hyperCKemia associated with CACNA1S variant.Eur J Neurol. 2018 Feb;25(2):417-419. doi: 10.1111/ene.13528. Epub 2017 Dec 26. Eur J Neurol. 2018. PMID: 29193480
-
A novel CACNA1S gene variant in a child with hypokalemic periodic paralysis: a case report and literature review.BMC Pediatr. 2023 Oct 2;23(1):500. doi: 10.1186/s12887-023-04326-1. BMC Pediatr. 2023. PMID: 37784084 Free PMC article. Review.
-
Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy.Acta Neuropathol. 2017 Apr;133(4):517-533. doi: 10.1007/s00401-016-1656-8. Epub 2016 Dec 23. Acta Neuropathol. 2017. PMID: 28012042
-
Neuromuscular symptoms in patients with RYR1-related malignant hyperthermia and rhabdomyolysis.Brain Commun. 2022 Nov 10;4(6):fcac292. doi: 10.1093/braincomms/fcac292. eCollection 2022. Brain Commun. 2022. PMID: 36751502 Free PMC article.
-
RYR1-Related Rhabdomyolysis: A Spectrum of Hypermetabolic States Due to Ryanodine Receptor Dysfunction.Curr Pharm Des. 2022;28(1):2-14. doi: 10.2174/1381612827666210804095300. Curr Pharm Des. 2022. PMID: 34348614 Review.
References
LinkOut - more resources
Full Text Sources
Research Materials
