

Genetic variants and phenotypic data curated for the CAGI6 intellectual disability panel challenge
- PMID: 40019509
- PMCID: PMC11976335
- DOI: 10.1007/s00439-025-02733-1
Genetic variants and phenotypic data curated for the CAGI6 intellectual disability panel challenge
Abstract
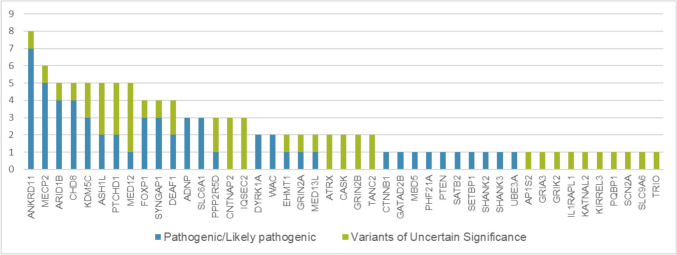
Neurodevelopmental disorders (NDDs) are common conditions including clinically diverse and genetically heterogeneous diseases, such as intellectual disability, autism spectrum disorders, and epilepsy. The intricate genetic underpinnings of NDDs pose a formidable challenge, given their multifaceted genetic architecture and heterogeneous clinical presentations. This work delves into the intricate interplay between genetic variants and phenotypic manifestations in neurodevelopmental disorders, presenting a dataset curated for the Critical Assessment of Genome Interpretation (CAGI6) ID Panel Challenge. The CAGI6 competition serves as a platform for evaluating the efficacy of computational methods in predicting phenotypic outcomes from genetic data. In this study, a targeted gene panel sequencing has been used to investigate the genetic causes of NDDs in a cohort of 415 paediatric patients. We identified 60 pathogenic and 49 likely pathogenic variants in 102 individuals that accounted for 25% of NDD cases in the cohort. The most mutated genes were ANKRD11, MECP2, ARID1B, ASH1L, CHD8, KDM5C, MED12 and PTCHD1 The majority of pathogenic variants were de novo, with some inherited from mildly affected parents. Loss-of-function variants were the most common type of pathogenic variant. In silico analysis tools were used to assess the potential impact of variants on splicing and structural/functional effects of missense variants. The study highlights the challenges in variant interpretation especially in cases with atypical phenotypic manifestations. Overall, this study provides valuable insights into the genetic causes of NDDs and emphasises the importance of understanding the underlying genetic factors for accurate diagnosis, and intervention development in neurodevelopmental conditions.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Conflict of interest: The authors declare no competing interests. Ethics approval and consent to participate: This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of University Hospital of Padua, Italy. According to approved protocols of each referring clinical centre, written informed consent was obtained from the probands or their legal representatives for specimen collection and genetic analysis. All individuals recruited provided informed consent for their participation in the study and publication of relevant findings.
Figures
References
-
- Aspromonte MC, Bellini M, Gasparini A, Carraro M, Bettella E, Polli R, Cesca F, Bigoni S, Boni S, Carlet O, Negrin S, Mammi I, Milani D, Peron A, Sartori S, Toldo I, Soli F, Turolla L, Stanzial F, Leonardi E (2019) Characterization of intellectual disability and autism comorbidity through gene panel sequencing. Hum Mutat 40(9):1346–1363. 10.1002/humu.23822 - PMC - PubMed
-
- Aspromonte MC, Del Conte A, Zhu S, Tan W, Shen Y, Zhang Y, Li Q, Wang MH, Babbi G, Bovo S, Martelli PL, Casadio R, Althagafi A, Toonsi S, Kulmanov M, Hoehndorf R, Katsonis P, Williams A, Lichtarge O, Leonardi E (2025) CAGI6 ID panel challenge: Assessment of phenotype and variant predictions in 415 children with neurodevelopmental disorders (NDDs). Human Genet. 10.1007/s00439-024-02722-w - PMC - PubMed
-
- Barbosa S, Greville-Heygate S, Bonnet M, Godwin A, Fagotto-Kaufmann C, Kajava AV, Laouteouet D, Mawby R, Wai HA, Dingemans AJM, Hehir-Kwa J, Willems M, Capri Y, Mehta SG, Cox H, Goudie D, Vansenne F, Turnpenny P, Vincent M, Baralle D (2020) Opposite Modulation of RAC1 by Mutations in TRIO Is Associated with Distinct, Domain-Specific Neurodevelopmental Disorders. The Am J Human Genet 106(3):338–355. 10.1016/j.ajhg.2020.01.018 - PMC - PubMed
MeSH terms
Grants and funding
- MUR CN00000041, C.F. 92315700283/European Union-NextGenerationEU through 'Italiadomani-PNRR' project CN - G.T.RNA SP. 7 project - National Center for Gene Therapy and Drugs based on RNA Technology (CN3)
- 3HP-HP-FPA ERN-01-2016/739516/European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability
LinkOut - more resources
Full Text Sources
