

Rapid brain tumor classification from sparse epigenomic data
- PMID: 40021833
- PMCID: PMC11922770
- DOI: 10.1038/s41591-024-03435-3
Rapid brain tumor classification from sparse epigenomic data
Abstract
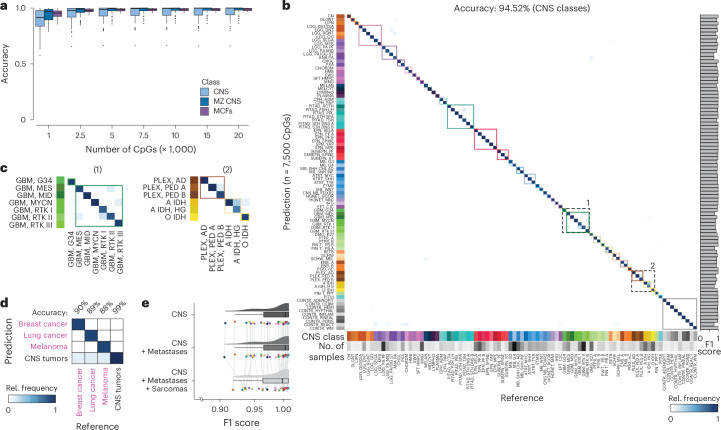
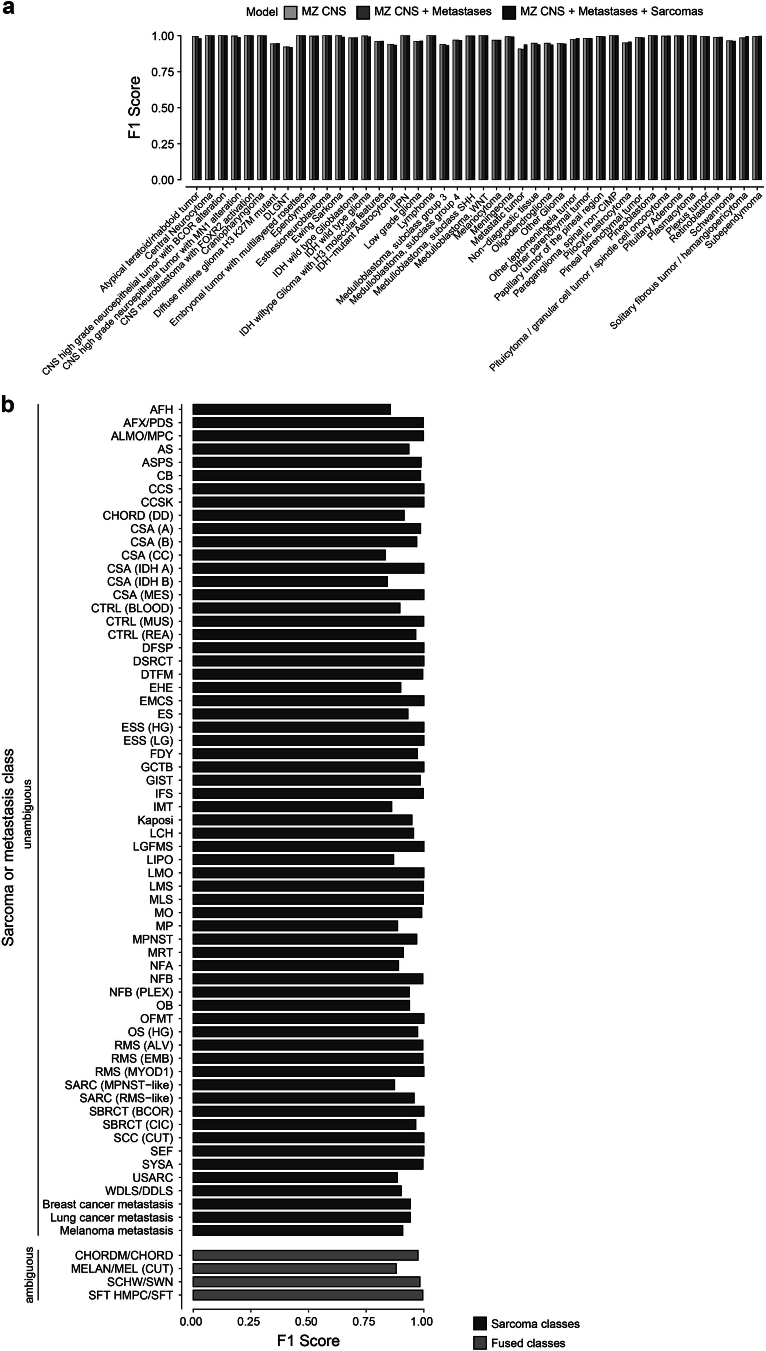
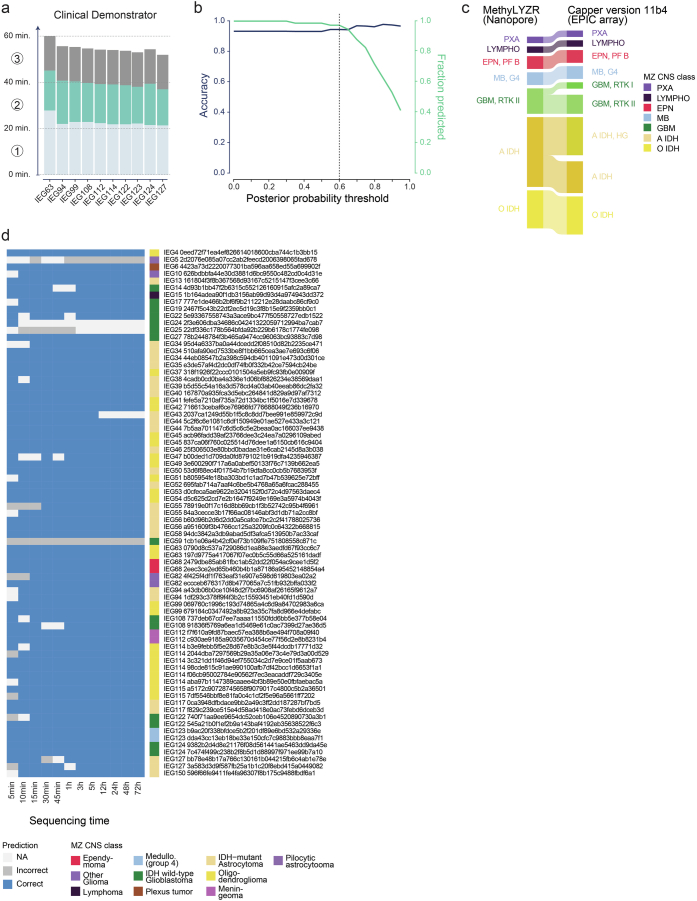
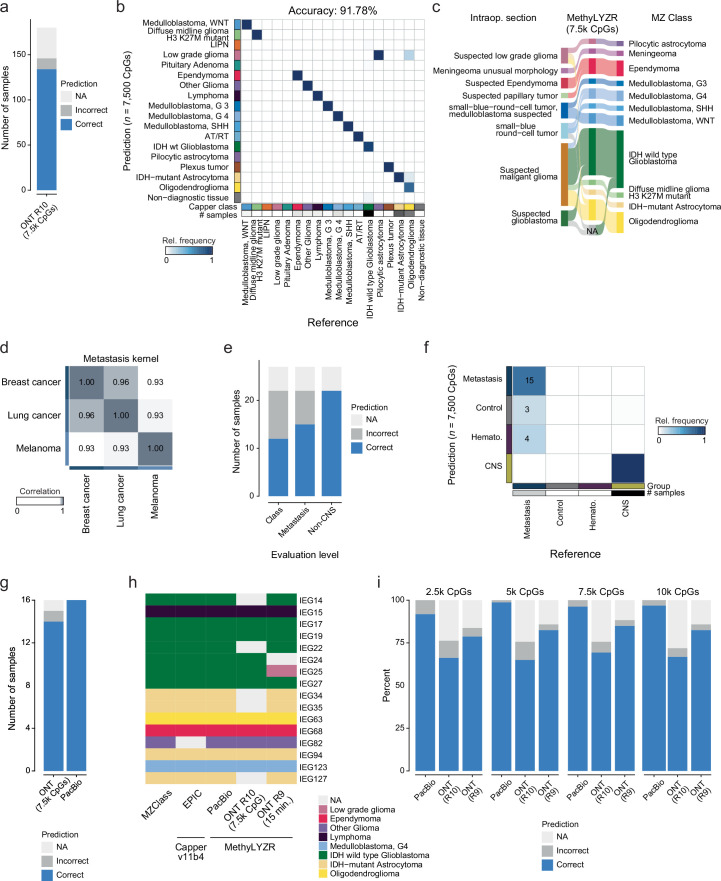
Although the intraoperative molecular diagnosis of the approximately 100 known brain tumor entities described to date has been a goal of neuropathology for the past decade, achieving this within a clinically relevant timeframe of under 1 h after biopsy collection remains elusive. Advances in third-generation sequencing have brought this goal closer, but established machine learning techniques rely on computationally intensive methods, making them impractical for live diagnostic workflows in clinical applications. Here we present MethyLYZR, a naive Bayesian framework enabling fully tractable, live classification of cancer epigenomes. For evaluation, we used nanopore sequencing to classify over 200 brain tumor samples, including 10 sequenced in a clinical setting next to the operating room, achieving highly accurate results within 15 min of sequencing. MethyLYZR can be run in parallel with an ongoing nanopore experiment with negligible computational overhead. Therefore, the only limiting factors for even faster time to results are DNA extraction time and the nanopore sequencer's maximum parallel throughput. Although more evidence from prospective studies is needed, our study suggests the potential applicability of MethyLYZR for live molecular classification of nervous system malignancies using nanopore sequencing not only for the neurosurgical intraoperative use case but also for other oncologic indications and the classification of tumors from cell-free DNA in liquid biopsies.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: F.-J.M., B.S., H.K., A.v.B., B.B. and C.R. filed a patent application (WO 2023/031485 A1) covering the development of MethyLYZR. S.Y. is a member of advisory boards and has received honoraria from Amgen, AstraZeneca, Bayer, Janssen, Roche and Servier. H.K. is a member of an expert panel and has received honoraria from Roche and Oxford Nanopore Technologies. The remaining authors declare no competing interests.
Figures









References
-
- Gal, A. A. & Cagle, P. T. The 100-year anniversary of the description of the frozen section procedure. JAMA294, 3135–3137 (2005). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
