

Noncanonical circRNA biogenesis driven by alpha and gamma herpesviruses
- PMID: 40033018
- PMCID: PMC12000468
- DOI: 10.1038/s44318-025-00398-0
Noncanonical circRNA biogenesis driven by alpha and gamma herpesviruses
Abstract
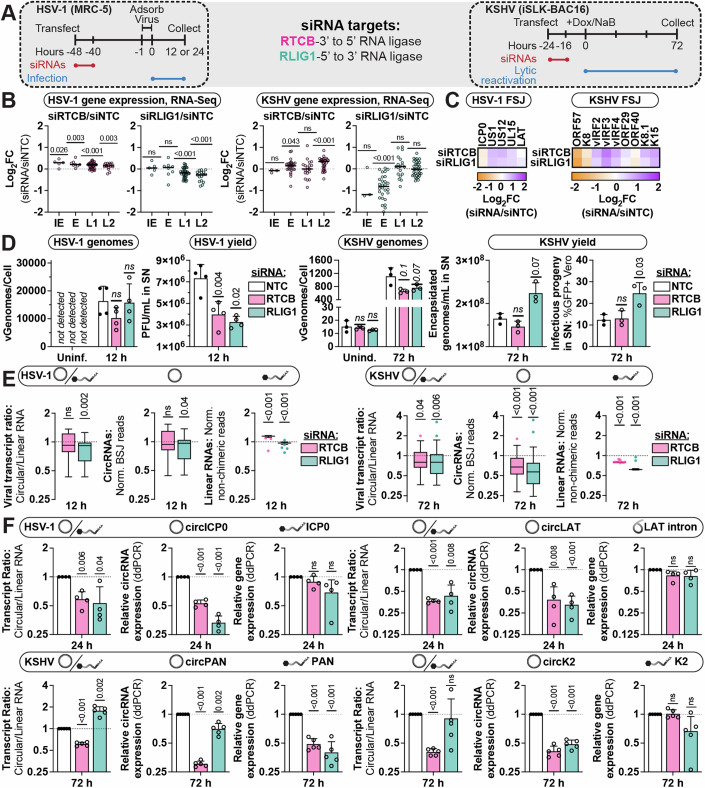
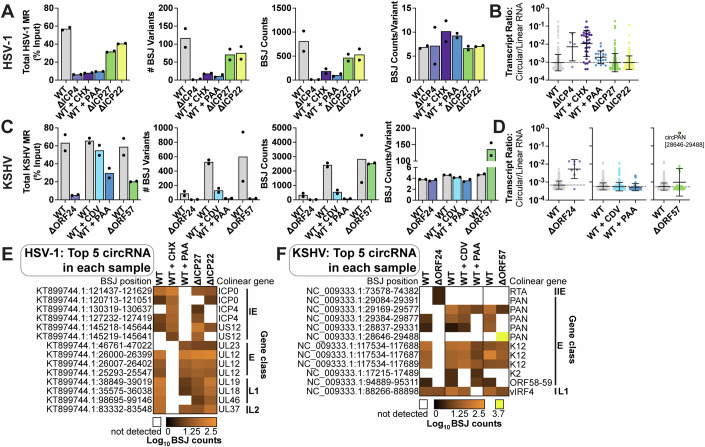
Herpesviruses require the host transcriptional machinery, inducing significant changes in gene expression to prioritize viral transcripts. We examined alpha- and gamma-herpesvirus alterations to a type of alternative splicing, namely circular RNA (circRNA) synthesis. We developed "Circrnas in Host And viRuses anaLysis pIpEline" (CHARLIE) to facilitate viral profiling. This method identified thousands of back-splicing variants, including circRNA common to lytic and latent phases of infection. Ours is the first report of Herpes Simplex Virus-1 circRNAs, including species derived from ICP0 and the latency-associated transcript. We characterized back-splicing cis- and trans-elements, and found viral circRNAs resistant to spliceosome perturbation and lacking canonical splice donor-acceptors. Subsequent loss-of-function studies of host RNA ligases (RTCB, RLIG1) revealed instances of decreased viral back splicing. Using eCLIP and 4sU-Sequencing, we determined that the KSHV RNA-binding protein, ORF57, enhanced synthesis for a subset of viral and host circRNAs. Our work explores unique splicing mechanisms driven by lytic infection, and identifies a class of transcripts with the potential to function in replication, persistence, or tumorigenesis.
Keywords: Alternative Splicing; Circular RNAs; Herpesviruses; RNA-Binding Proteins.
© 2025. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
Disclosure and competing interests statement. The authors declare no competing interests.
Figures
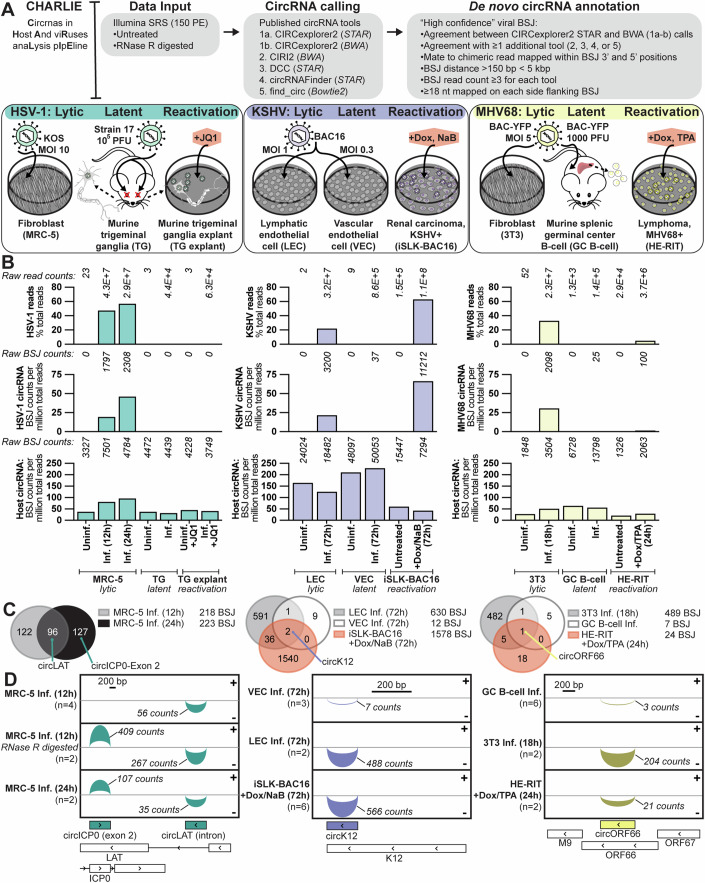
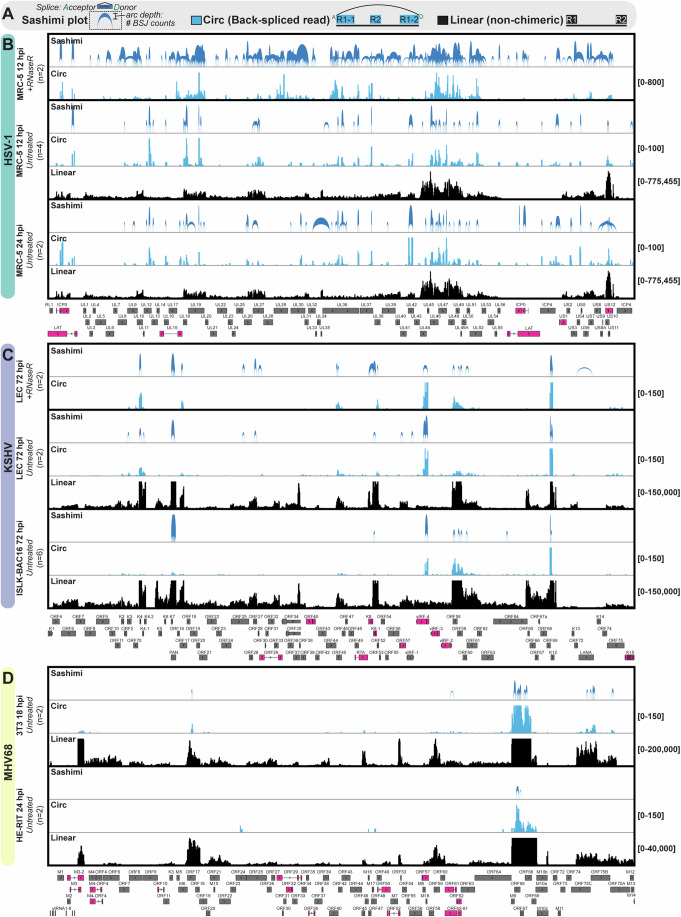
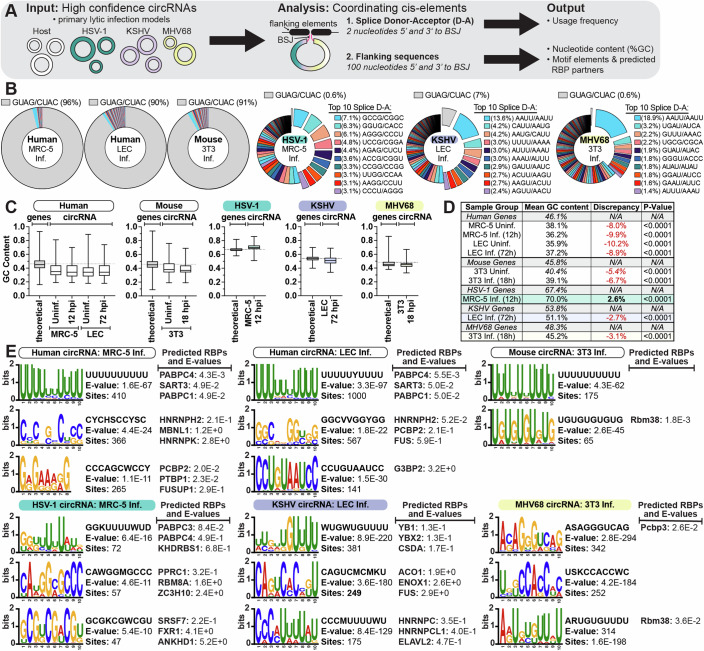
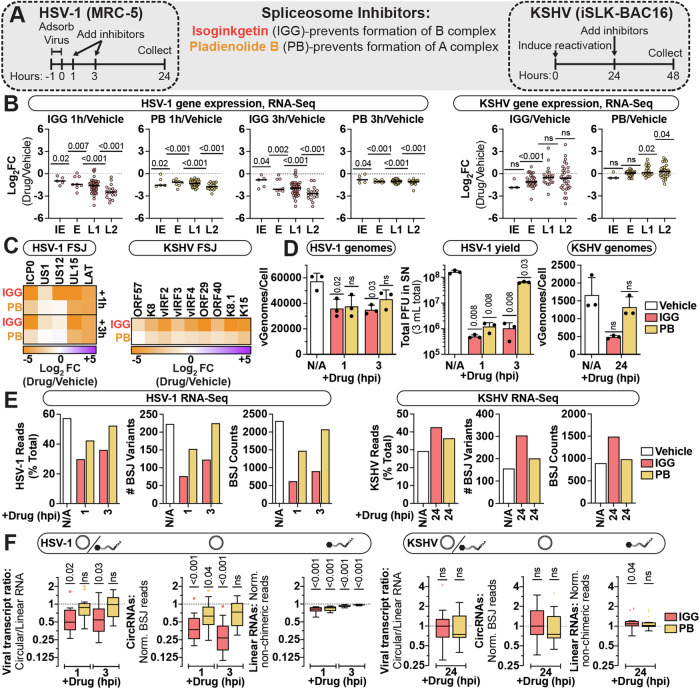






References
-
- Alwine JC, Steinhart WL, Hill CW (1974) Transcription of herpes simplex type 1 DNA in nuclei isolated from infected HEp-2 and KB cells. Virology 60:302–307 - PubMed
-
- Arias C, Weisburd B, Stern-Ginossar N, Mercier A, Madrid AS, Bellare P, Holdorf M, Weissman JS, Ganem D (2014) KSHV 2.0: a comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog 10:e1003847 - PMC - PubMed
-
- Ashwal-Fluss R, Meyer M, Pamudurti Nagarjuna R, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S (2014) circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 56:55–66 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
