

Systematic analysis of specificities and flanking sequence preferences of bacterial DNA-(cytosine C5)-methyltransferases reveals mechanisms of enzyme- and sequence-specific DNA readout
- PMID: 40037710
- PMCID: PMC11879396
- DOI: 10.1093/nar/gkaf126
Systematic analysis of specificities and flanking sequence preferences of bacterial DNA-(cytosine C5)-methyltransferases reveals mechanisms of enzyme- and sequence-specific DNA readout
Abstract
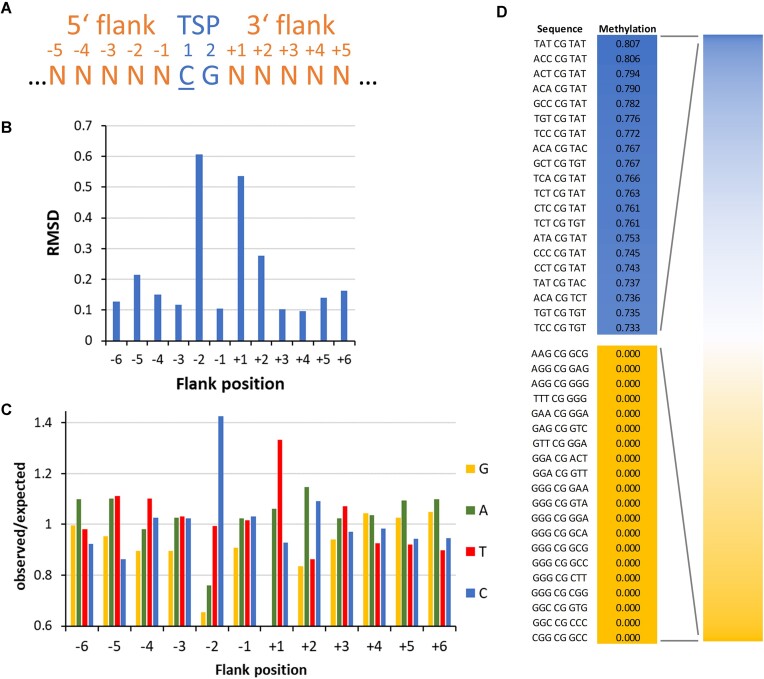
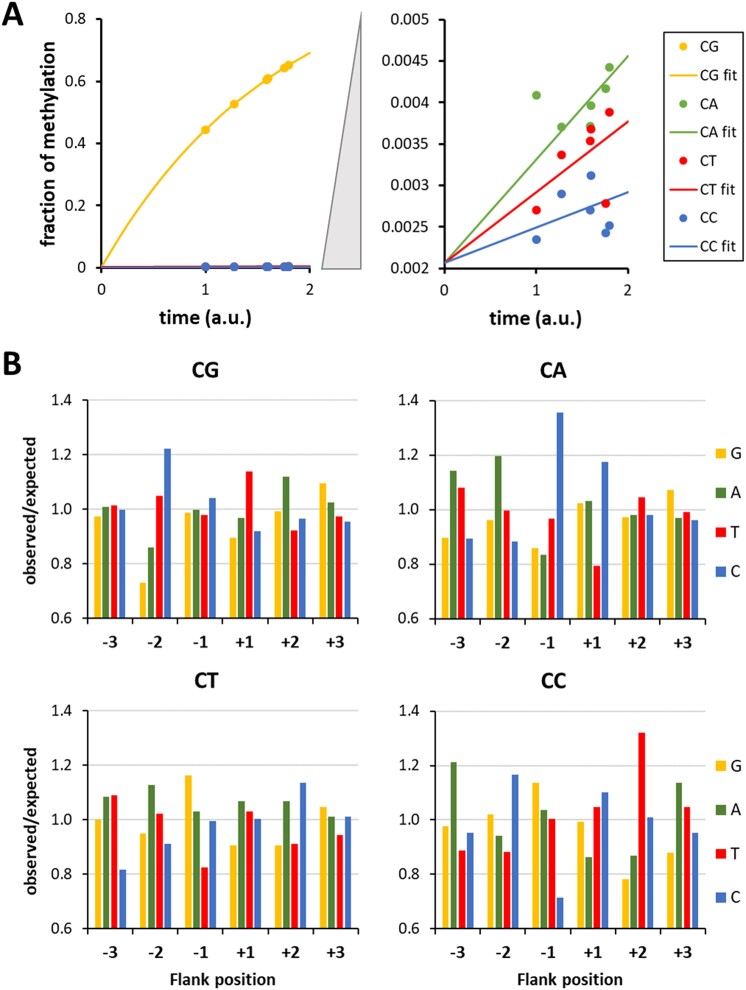
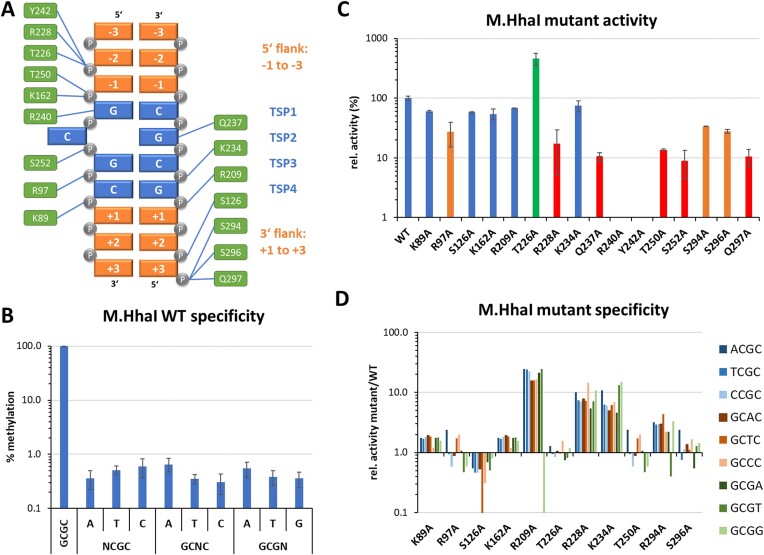
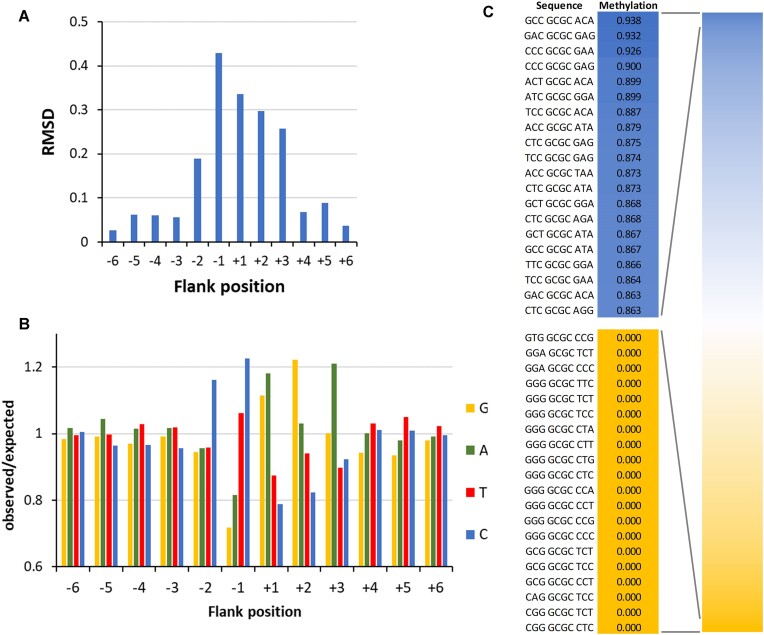
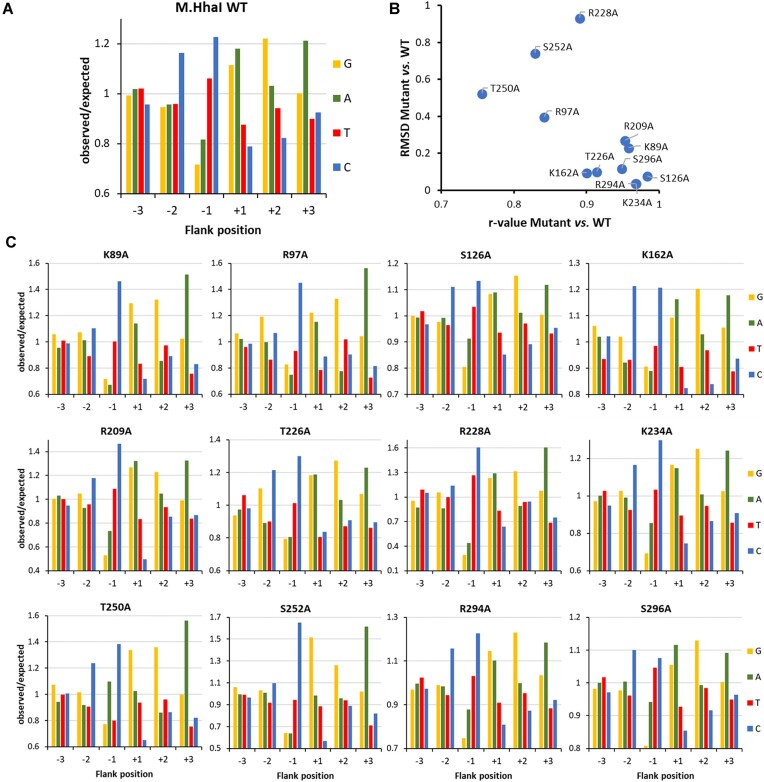
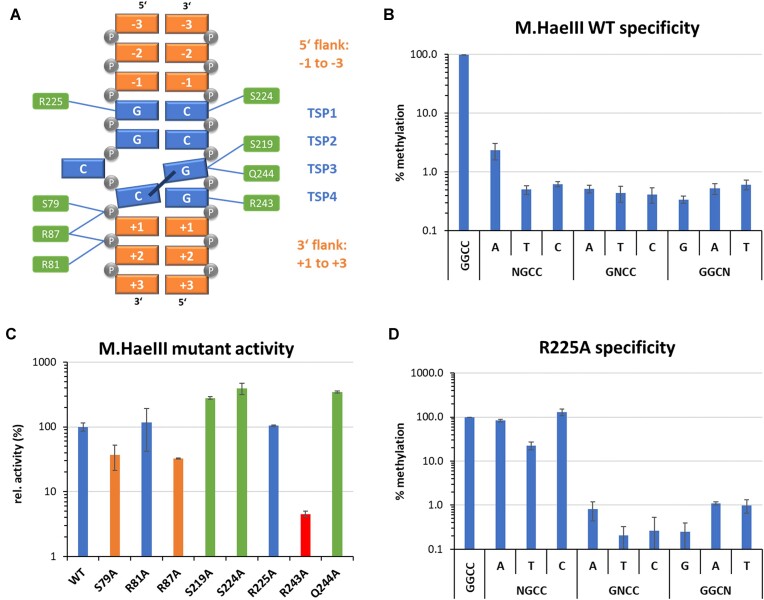
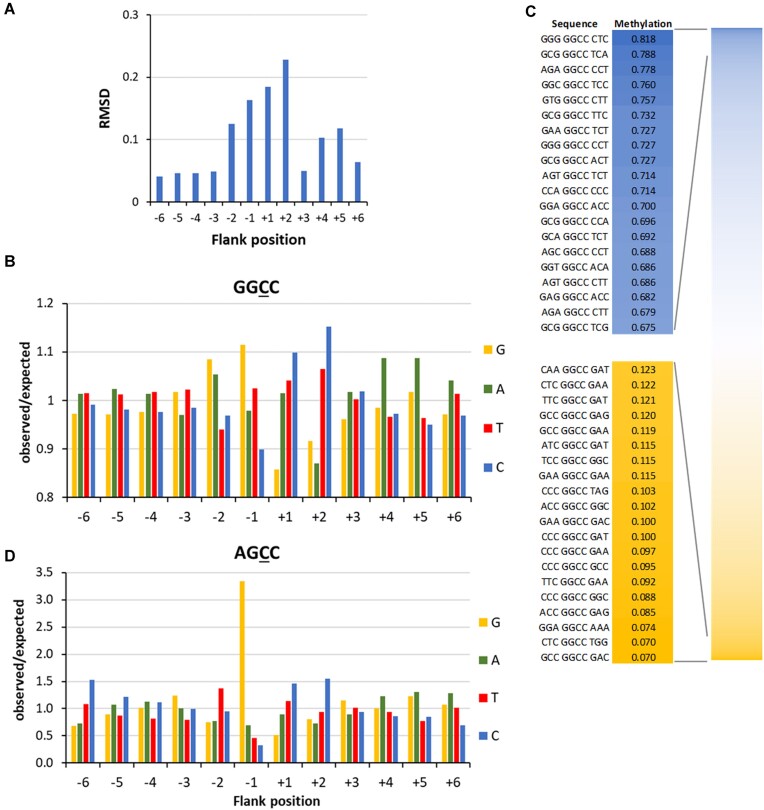
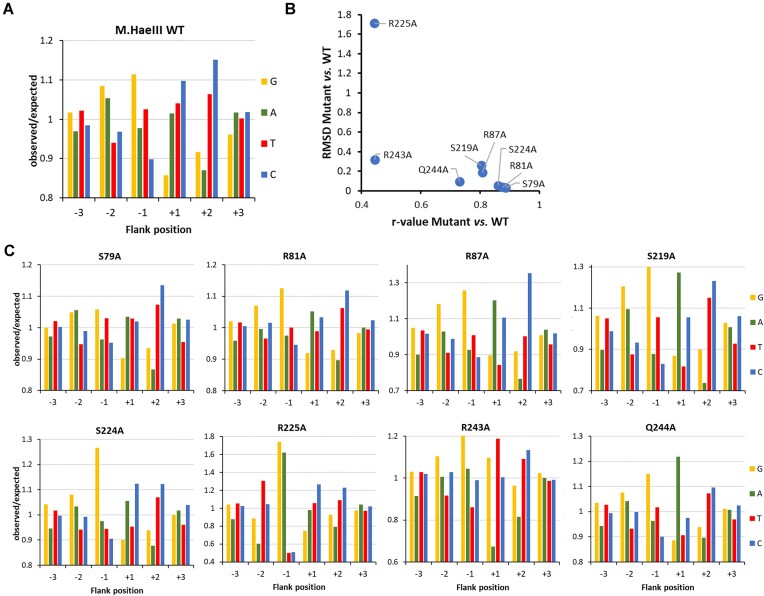
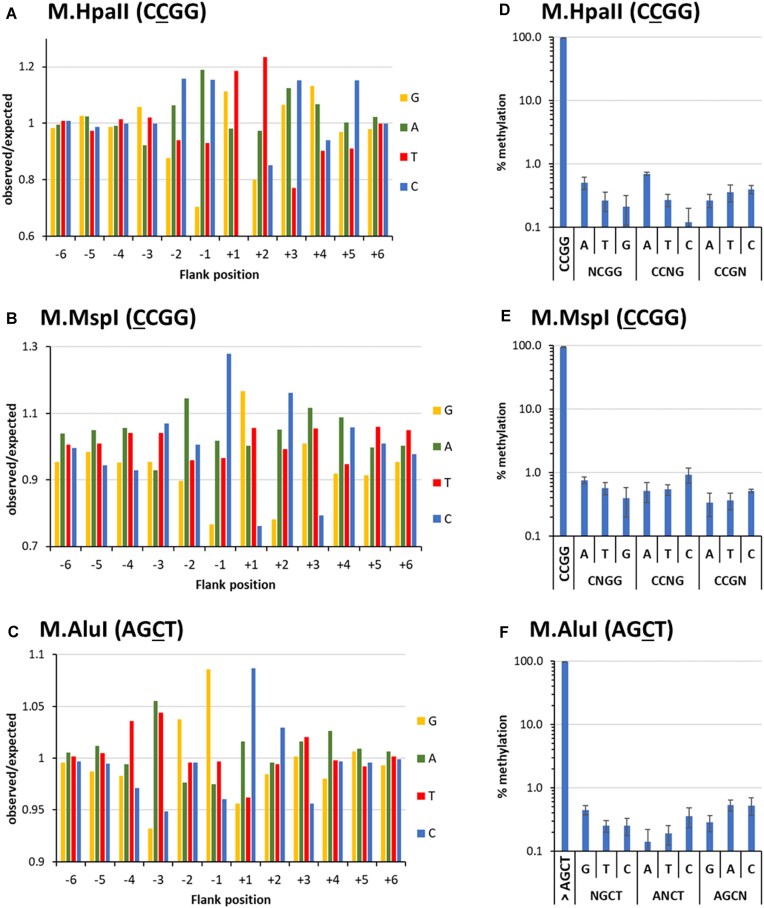
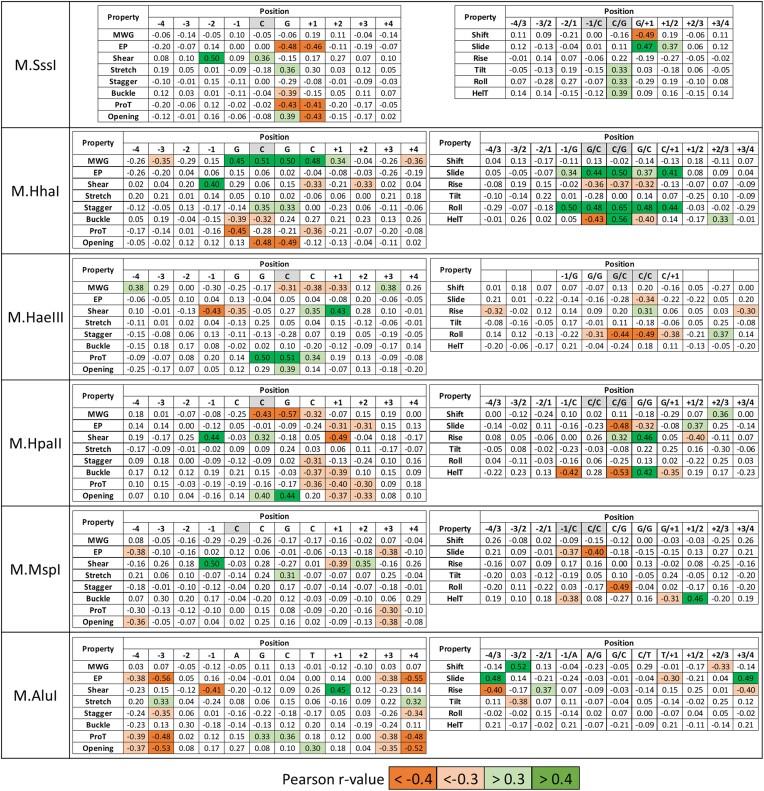
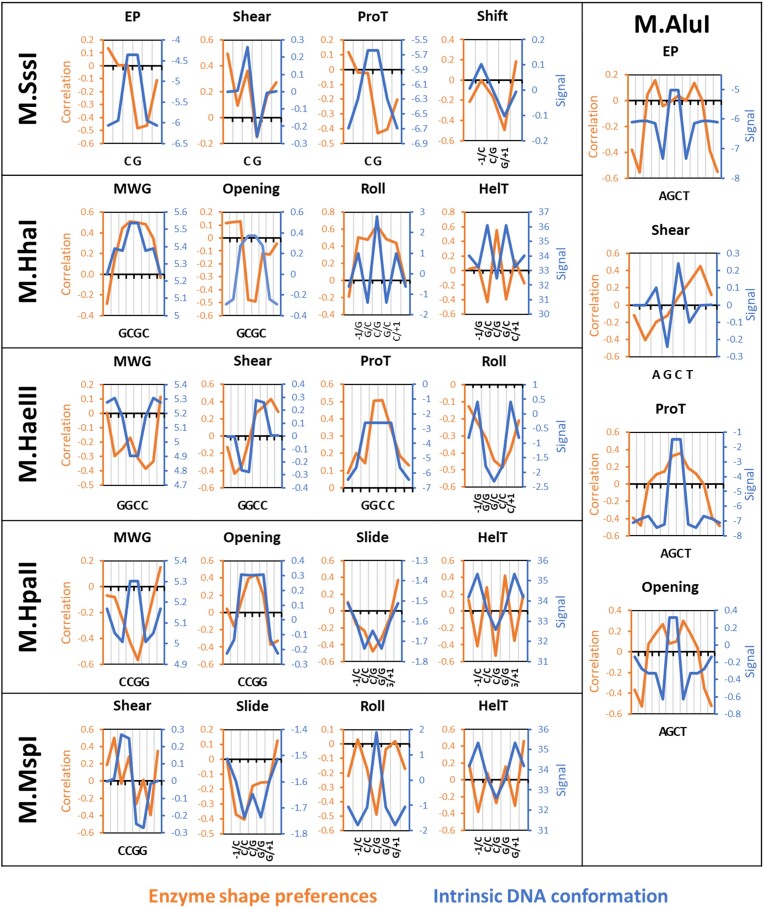
DNA-(cytosine C5)-methyltransferases (MTases) represent a large group of evolutionary related enzymes with specific DNA interaction. We systematically investigated the specificity and flanking sequence preferences of six bacterial enzymes of this class and many MTase mutants. We observed high (>1000-fold) target sequence specificity reflecting strong evolutionary pressure against unspecific DNA methylation. Strong flanking sequence preferences (∼100-fold) were observed which changed for methylation of near-cognate sites suggesting that the DNA structures in the transition states of the methylation of these sites differ. Mutation of amino acids involved in DNA contacts led to local changes of specificity and flanking sequence preferences, but also global effects indicating that larger conformational changes occur upon transition state formation. Based on these findings, we conclude that the transition state of the DNA methylation reaction precedes the covalent enzyme-DNA complex conformations with flipped target base that are resolved in structural studies. Moreover, our data suggest that alternative catalytically active conformations exist whose occupancy is modulated by enzyme-DNA contacts. Sequence dependent DNA shape analyses suggest that MTase flanking sequence preferences are caused by flanking sequence dependent modulation of the DNA conformation. Likely, many of these findings are transferable to other DNA MTases and DNA interacting proteins.
© The Author(s) 2025. Published by Oxford University Press on behalf of Nucleic Acids Research.
Conflict of interest statement
None declared.
Figures
Similar articles
-
Distal structural elements coordinate a conserved base flipping network.Biochemistry. 2013 Mar 12;52(10):1669-76. doi: 10.1021/bi301284f. Epub 2013 Feb 27. Biochemistry. 2013. PMID: 23409802
-
Enzymatic C5-cytosine methylation of DNA: mechanistic implications of new crystal structures for HhaL methyltransferase-DNA-AdoHcy complexes.J Mol Biol. 1996 Sep 6;261(5):634-45. doi: 10.1006/jmbi.1996.0489. J Mol Biol. 1996. PMID: 8800212
-
The fission yeast gene pmt1+ encodes a DNA methyltransferase homologue.Nucleic Acids Res. 1995 Jan 25;23(2):203-10. doi: 10.1093/nar/23.2.203. Nucleic Acids Res. 1995. PMID: 7862522 Free PMC article.
-
Deep Enzymology Studies on DNA Methyltransferases Reveal Novel Connections between Flanking Sequences and Enzyme Activity.J Mol Biol. 2021 Sep 17;433(19):167186. doi: 10.1016/j.jmb.2021.167186. Epub 2021 Aug 8. J Mol Biol. 2021. PMID: 34375615 Review.
-
Structure, function, and mechanism of HhaI DNA methyltransferases.Crit Rev Biochem Mol Biol. 2002;37(3):167-97. doi: 10.1080/10409230290771492. Crit Rev Biochem Mol Biol. 2002. PMID: 12139442 Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
