

AAV9-mediated MYBPC3 gene therapy with optimized expression cassette enhances cardiac function and survival in MYBPC3 cardiomyopathy models
- PMID: 40038304
- PMCID: PMC11880196
- DOI: 10.1038/s41467-025-57481-7
AAV9-mediated MYBPC3 gene therapy with optimized expression cassette enhances cardiac function and survival in MYBPC3 cardiomyopathy models
Abstract
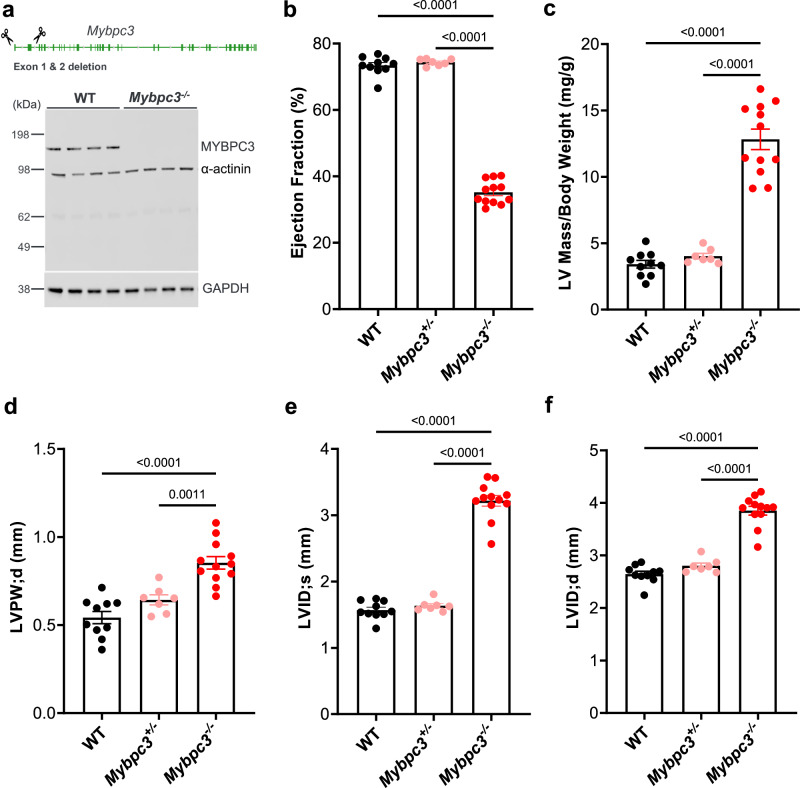
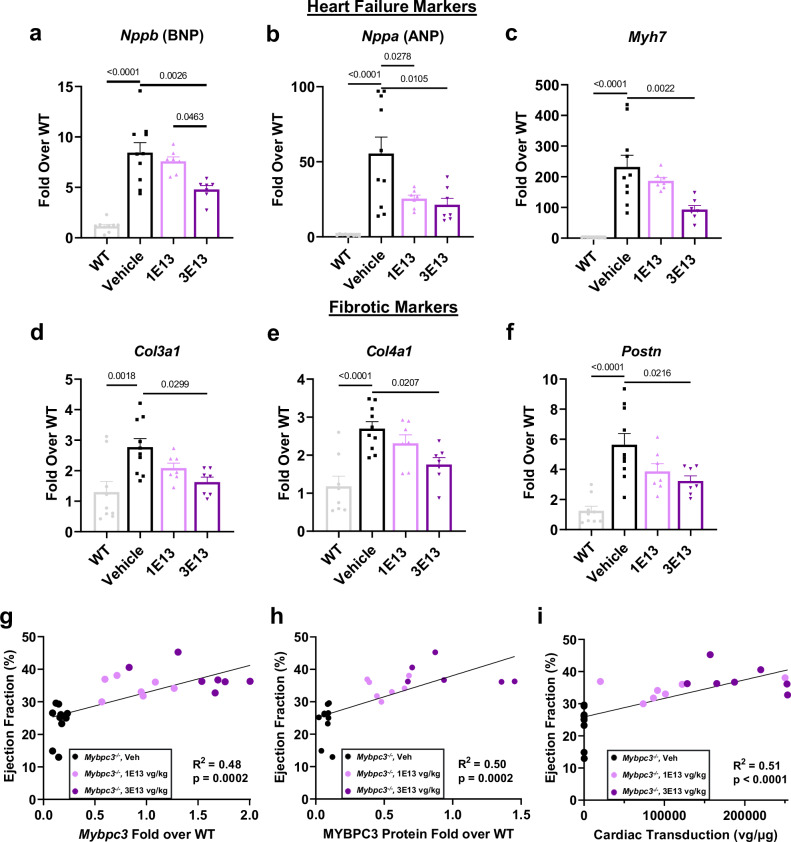
Hypertrophic cardiomyopathy (HCM) affects approximately 600,000 people in the United States. Loss-of-function mutations in Myosin Binding Protein C3, MYBPC3, are the most common genetic cause of HCM, with the majority of mutations resulting in haploinsufficiency. To restore cardiac MYBPC3, we use an adeno-associated virus (AAV9) vector and engineer an optimized expression cassette with a minimal promoter and cis-regulatory elements (TN-201) to enhance packaging efficiency and cardiomyocyte expression. Rather than simply preventing cardiac dysfunction preclinically, we demonstrate in a symptomatic MYBPC3-deficient murine model the ability of AAV gene therapy to reverse cardiac hypertrophy and systolic dysfunction, improve diastolic dysfunction, and prolong survival. Dose-ranging efficacy studies exhibit restoration of wild-type MYBPC3 protein levels and saturation of cardiac improvement at the clinically relevant dose of 3E13 vg/kg, outperforming a previously published construct. These findings suggest that TN-201 may offer therapeutic benefits in MYBPC3-associated cardiomyopathy, pending further validation in clinical settings.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors of this publication declare the following competing interests: All authors of this publication were employed by Tenaya Therapeutics at the time of the study and hold or held equity in Tenaya Therapeutics. L.M.L. is an inventor on published U.S. Patent No.: US 2023/0372541 A1 held by Tenaya Therapeutics that covers gene therapy cassettes for treating heart disease.
Figures







References
-
- Norrish, G. & Kaski, J. P. The risk of sudden death in children with hypertrophic cardiomyopathy. Heart Fail Clin.18, 9–18 (2022). - PubMed
-
- Maron, B. J. & Maron, M. S. Hypertrophic cardiomyopathy. Lancet381, 242–255 (2013). - PubMed
-
- Maron, B. J., Maron, M. S. & Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J. Am. Coll. Cardiol.60, 705–715 (2012). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
